 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 47(2); 2013 > Article
-
Case Study
Congenital Peribronchial Myofibroblastic Tumor: A Case Study and Literature Review - Yuil Kim, Ha Young Park, Junhun Cho, Joungho Han, Eun Yoon Cho
-
Korean Journal of Pathology 2013;47(2):172-176.
DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.2.172
Published online: April 24, 2013
Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- Corresponding Author: Eun Yoon Cho, M.D. Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea. Tel: +82-2-3410-2800, Fax: +82-2-3410-0025, eunyoon.cho@samsung.com
© 2013 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Congenital peribronchial myofibroblastic tumor (CPMT) is a benign pulmonary spindle cell neoplasm of intrauterine and perinatal period, which is thought to arise from primitive peribronchial mesenchyme. We present a case detected incidentally in a one-month-old infant. The solid and partially necrotic tumor involved the right middle and lower lobes of the lung with extension to the diaphragm. Histologically, the tumor was composed of fasciculated monotonous spindle cells, proliferating peribronchiolar cartilage and round cells with rich vasculature, and high mitotic activity was identified in the round cell area. Immunohistochemical and electron microscopic studies showed that the spindle cells were myofibroblastic in phenotype. Although the tumor showed several malignant pathological features, recurrence was not observed in the two-year follow-up period, consistent with the benign clinical behavior of CPMT.
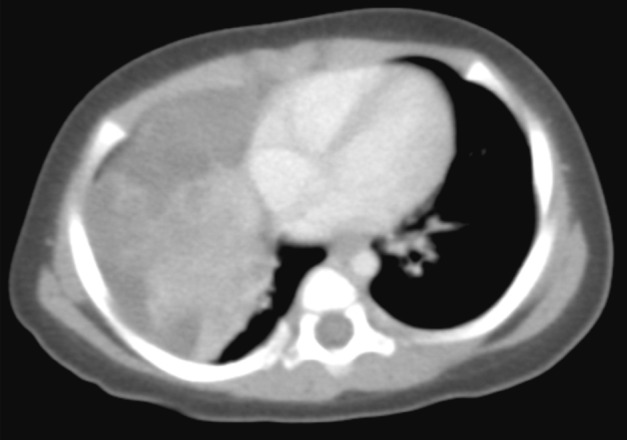
- The patient was a female infant born at 38 weeks gestation by Caesarian section without prenatal complications. At one month of age, a right lung lesion was discovered on a chest X-ray taken for evaluation of prolonged diarrhea. Subsequent computed tomography scan at 47 days of age showed a large solid and cystic mass involving the right middle lobe and the right lower lobe of the lung (Fig. 1). With the radiologic impression of mesenchymal tumor or pleuropulmonary blastoma, the baby presented to our institution for further management. At admission, the baby at 80 days of age had no respiratory or gastrointestinal symptoms, and no abnormality was noted on laboratory tests. Four days later, a thoracotomy with right middle and lower bilobectomy was performed to remove the lung mass. The mass extended into the diaphragm, but it was dissectible without causing a defect in the diaphragm. The patient received no further treatment after the surgery and had been healthy without evidence of recurrence for two years until lost to follow-up.
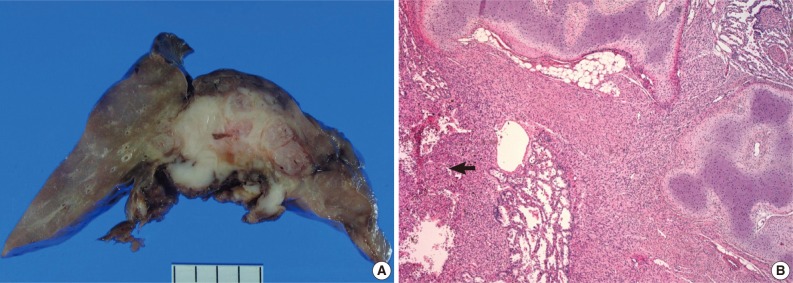
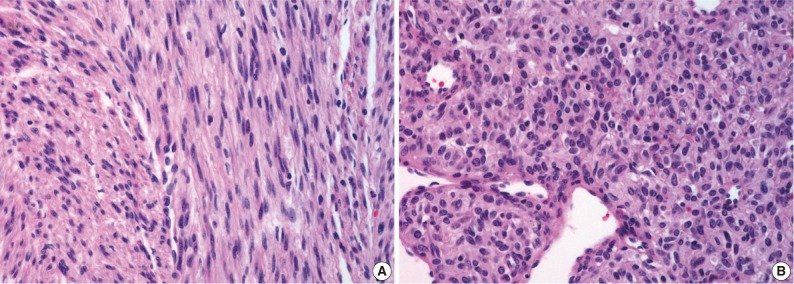
- On gross examination, the lung showed a partially necrotic solid mass, which measured 5.5×5×4 cm out of the 11×6×5 cm-sized bilobectomy specimen. The cut surface of the solid portion was white to pink with a trabeculated area and cartilaginous foci (Fig. 2A). The border with the lung parenchyma was indistinct in some areas. Microscopically, the tumor was in irregular shape consisting of three components (Fig. 2B). Fascicles of monotonous spindle cells constituted the largest portion (Fig. 3A). The cords and nodules of spindle cells infiltrated the lung parenchyma at the tumor boundary. The second component was broad irregular plates of mature cartilage, which were associated with airways. The third component was sheets of round to polygonal cells enriched with vascular channels, comparable to a hemangiopericytoma-like pattern (Fig. 3B). These round cells were distributed near the foci of necrosis/hemorrhagic degeneration with gradual transformation from spindle cells. Higher cellularity with a moderate degree of nuclear pleomorphism was observed in the round cell area, in which the mitosis was counted to be as many as eight per ten high power fields (hpf). Additionally, rare foci of adipose tissue were observed at the periphery of the tumor (Fig. 2B). The bronchial resection margin and the visceral pleura were involved by the tumor. The background lung parenchyma was unremarkable.
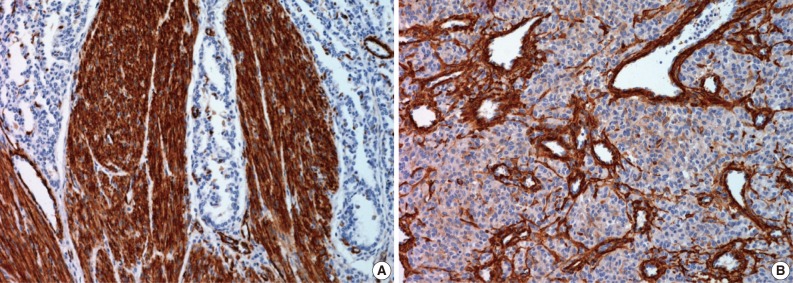
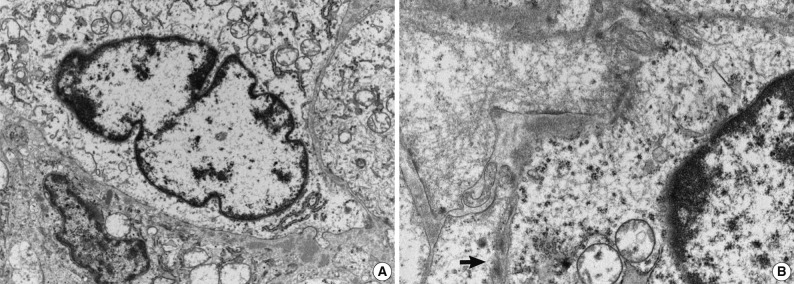
- Immunohistochemical study showed that the spindle cell and the round cell were positive for vimentin, but only the spindle cell was strongly reactive for smooth muscle actin (Fig. 4). Ki-67 staining was positive in about 1% and 5% in the spindle cell and round cell areas, respectively. The chondrocytes in the cartilage component were positive for S-100. Desmin, cytokeratin, epithelial membrane antigen, and factor VIII were negative in the tumor. On electron microscopic examination of spindle cell areas (Fig. 5), uniform oval to polygonal cells with smooth cell borders and occasional primitive intercellular junctions were observed. The cytoplasm was abundant and contained rough endoplasmic reticulum of variable density, mitochondria, free ribosomes and a few foci of dense bodies with cytoplasmic filaments. Pinocytotic vesicles or basal lamina were also observed. These findings indicate primitive mesenchymal cells with partial myofibroblastic differentiation, which is compatible with the expression of vimentin and smooth muscle actin.
CASE REPORT
- The microscopic pattern of CPMT spans from that of uniform spindle cells4,12,13 to that harboring peribronchial cartilage proliferation6,9,14-16 and/or having a hemangiopericytoma-like area.2,10,17 The phenotype of the tumor cells has been related to that of myofibroblasts or fibroblasts, with variable reactivity to smooth muscle actin or muscle specific actin1,2,10,12-17 and the variable presence of cytoplasmic filaments in electron microscopy. 1,2,4,7,15,17 McGinnis et al.1 have proposed that the tumor arises from primitive peribronchial mesenchyme that may differentiate to form smooth muscle and cartilage. A recent account of CPMT provided supporting evidence for this notion by demonstrating the differentiating tendency in the histology of the same tumor obtained by fetal surgery and postnatal surgery.16 Over the four-month interval of observation, the authors noted decreased cellularity of the spindle cell component and increased proportion and maturation of the cartilage component of the tumor.16 The current case is unique in presenting a tumor in an infant, while all previous reports described tumors from the perinatal period (Table 1). The prominence of the cartilage component in the current case is also supportive of the proposed differentiating potential of the tumor.
- Since CPMT has been known to manifest certain features of malignant neoplasm, such as frequent mitosis (greater than 10/10 hpf2,5,7), necrosis,2,12,13,17 and infiltrative growth, some CPMT cases have been reported as malignant entities, such as bronchopulmonary fibrosarcoma2,3 and bronchopulmonary leiomyosarcoma.4,5 However, this neoplasm is benign, as it has not recurred or metastasized in patients surviving through the tumor-removal operation, even with positive surgical resection margins (current case and Huppmann et al.16). The tumor has been mostly associated with hydrops fetalis and neonatal respiratory distress, which has led to in utero and perinatal morbidity and mortality. The current case is among the rare instances3,10 in which the patient was asymptomatic at detection. Cytogenetic analysis of CPMT was conducted in a few reports,1,16,17 and complex chromosomal rearrangements were shown in one.17
- The differential diagnosis of CPMT has been extensively discussed in previous reports.1,10,14 Two entities deserve recapitulation for the current case. The solid, type III pleuropulmonary blastoma may show sheets of spindle cells, chondroid foci and necrosis. Unlike CPMT, pleomorphic and anaplastic cells are characteristic, and the clinical course is that of malignancy.18 Bronchopulmonary fibrosarcoma is a low-grade malignancy, which presents as an endobronchial or intraparenchymal mass consisting of bundled spindle cells. Among 30 pediatric cases reported in the literature,2,19,20 four of five neonatal cases have been reclassified as CPMT.1,13 The recent, fifth neonatal case20 may also be considered a CPMT as it was composed of monotonous spindle cells with a high mitotic rate (20/10 hpf) and had not recurred during the 16-month follow-up. It has been suggested that CPMT and pediatric bronchopulmonary fibrosarcoma may be in a spectrum of the same neoplasm.1,14
- As CPMT shows good prognosis once adequately resected, it is important not to misdiagnose it as a malignant neoplasm, thereby subjecting the patient to unneeded chemotherapy or radiation therapy. The current case is notable in that it may be one of the most malignant-appearing CPMT cases reported, as it was characterized by a high mitotic rate, necrosis and invasion of the diaphragm.
DISCUSSION
Acknowledgments
Acknowledgments
- 1. McGinnis M, Jacobs G, el-Naggar A, Redline RW. Congenital peribronchial myofibroblastic tumor (so-called "congenital leiomyosarcoma"): a distinct neonatal lung lesion associated with nonimmune hydrops fetalis. Mod Pathol 1993; 6: 487-492. PubMed
- 2. Pettinato G, Manivel JC, Saldana MJ, Peyser J, Dehner LP. Primary bronchopulmonary fibrosarcoma of childhood and adolescence: reassessment of a low-grade malignancy. Clinicopathologic study of five cases and review of the literature. Hum Pathol 1989; 20: 463-471. ArticlePubMed
- 3. Robb D. A case of neonatal fibrosarcoma of lung. Br J Surg 1958; 46: 173-174. ArticlePubMedPDF
- 4. Jimenez JF, Uthman EO, Townsend JW, Gloster ES, Seibert JJ. Primary bronchopulmonary leiomyosarcoma in childhood. Arch Pathol Lab Med 1986; 110: 348-351. PubMed
- 5. Guccion JG, Rosen SH. Bronchopulmonary leiomyosarcoma and fibrosarcoma: a study of 32 cases and review of the literature. Cancer 1972; 30: 836-847. ArticlePubMed
- 6. Khong TY, Keeling JW. Massive congenital mesenchymal malformation of the lung: another cause of non-immune hydrops. Histopathology 1990; 16: 609-611. ArticlePubMed
- 7. Warren JS, Seo IS, Mirkin LD. Massive congenital mesenchymal malformation of the lung: a case report with ultrastructural study. Pediatr Pathol 1985; 3: 321-328. ArticlePubMed
- 8. Haller JO, Kauffman SL, Kassner EG. Congenital mesenchymal tumour of the lung. Br J Radiol 1977; 50: 217-219. ArticlePubMed
- 9. Jones CJ. Unusual hamartoma of the lung in a newborn infant. Arch Pathol (Chic) 1949; 48: 150-154. PubMed
- 10. Kuhnen C, Harms D, Niessen KH, Diehm T, Müller KM. Congenital pulmonary fibrosarcoma: differential diagnosis of infantile pulmonary spindle cell tumors. Pathologe 2001; 22: 151-156. PubMed
- 11. An J, Han J, Ahn KM, Kim J, Choi YS. Primary neoplasms of the lung in children and adolescents: 22 cases from a single institute. J Lung Cancer 2009; 8: 103-110. Article
- 12. Horikoshi T, Kikuchi A, Matsumoto Y, et al. Fetal hydrops associated with congenital pulmonary myofibroblastic tumor. J Obstet Gynaecol Res 2005; 31: 552-555. ArticlePubMed
- 13. Reiss A, Goldberg Y, Monichor M, Drugan A. Congenital pulmonary myofibroblastic tumor: pathology and prenatal sonographic appearance. Prenat Diagn 2005; 25: 1064-1066. ArticlePubMed
- 14. Dishop MK. Congenital pulmonary myofibroblastic tumor [Internet]. 2008; cite 2012 Jul 5. Augusta: United States and Canadian Academy of Pathology, Available from: http://www.uscap.org/site~/97th/specpulmh4v.htm.
- 15. de Noronha L, Cecílio WA, da Silva TF, Maggio EM, Serapião MJ. Congenital peribronchial myofibroblastic tumor: a case report. Pediatr Dev Pathol 2010; 13: 243-246. ArticlePubMedPDF
- 16. Huppmann AR, Coffin CM, Hoot AC, Kahwash S, Pawel BR. Congenital peribronchial myofibroblastic tumor: comparison of fetal and postnatal morphology. Pediatr Dev Pathol 2011; 14: 124-129. ArticlePubMedPDF
- 17. Alobeid B, Beneck D, Sreekantaiah C, Abbi RK, Slim MS. Congenital pulmonary myofibroblastic tumor: a case report with cytogenetic analysis and review of the literature. Am J Surg Pathol 1997; 21: 610-614. ArticlePubMed
- 18. Dishop MK, Kuruvilla S. Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children's hospital. Arch Pathol Lab Med 2008; 132: 1079-1103. ArticlePubMedPDF
- 19. Savas C, Candir O, Ozguner F. Acute respiratory distress due to fibrosarcoma of the carina in a child. Pediatr Pulmonol 2004; 38: 355-357. ArticlePubMed
- 20. Sahin D, Koç N, Etker S, Güven S, Canpolat C. Congenital pulmonary fibrosarcoma in a newborn with hypoglycemia and respiratory distress: case report. Turk J Pediatr 2010; 52: 325-329. PubMed
REFERENCES
| Reference | Sex/Agea | Outcome | Complication | Original diagnosis | Adjunct study | Cartilage | Necrosis | Round cell | Mitosis |
|---|---|---|---|---|---|---|---|---|---|
| Jones [9] | F/28 wk | Died at 1 hr | RD | Hamartoma | - | + | NS | NS | NS |
| Robb [3] | M/NS | Well at 10 mo | None | Fibrosarcoma | - | NS | NS | + | Not numerous |
| Guccion and Rosen [5] | M/NS | Died shortly after birth | NS | Leiomyosarcoma | - | NS | NS | NS | 15/10 hpf |
| Haller et al. [8] | F/40 wk | Died at 8 hr | H, RD | Mesenchymal tumor | - | NS | NS | + | None |
| Warren et al. [7] | F/30-33 wk | Intraoperative death | PHA, RD | Mesenchymal malformation | EM | Entrappedb | NS | NS | 1-5/hpf |
| Jimenez et al. [4] | M/36 wk | Well at 34 mo | H, PHA, RD | Leiomyosarcoma | EM | NS | NS | NS | Occasional |
| Pettinato et al. [2] | M/NS | Well at 6 yr | PE, RD | Fibrosarcoma | IHC, EM | Entrapped | + | + | 8-12/10 hpf |
| M/NS | Well at 3 mo | PE, RD | |||||||
| Khong and Keeling [6] | M/27 wk | Termination | H, PHA | Mesenchymal | - | + | NS | NS | Frequent |
| McGinnis et al. [1] | M/33 wk | Intraoperative death | H, PHA, RD | CPMT | IHC, EM, flow cytometry | + | - | NS | 1/10 hpf |
| Alobeid et al. [17] | F/35 wk | Well at 12 mo | RD | CPMT | IHC, EM, karyotyping | Entrapped | + | NS | 0-3/10 hpf |
| Kuhnen et al. [10] | NS/NS | Well at 8 mo | None | Fibrosarcoma | IHC | + | - | NS | 1/10 hpf |
| Reiss et al. [13] | M/24 wk | Termination | NS | CPMT | IHC | - | + | NS | 0-4/10 hpf |
| Horikoshi et al. [12] | M/30 wk | Intrauterine death | H, PE | CPMT | IHC | - | + | NS | NS |
| Dishop [14] | M/28 wk | Died 24 hr | H, RD | CPMT | IHC | + | - | NS | Occasional |
| de Noronha et al. [15] | M/24 wk | Intrauterine death | H, PHA | CPMT | IHC, EM | + | NS | NS | Frequent |
| Huppmann et al. [16] | M/23 wk, 6 wk postnatal | Well at 6.5 yr | H, RD | CPMT | IHC, karyotyping | + | - | NS | Brisk/occasional |
| Current case | F/1 mo | Well at 2 yr | None | CPMT | IHC, EM | + | + | + | 8/10 hpf |
CPMT, congenital peribronchial myofibroblastic tumor; F, female; RD, respiratory distress; NS, not specified; M, male; hpf, high power fields; H, hydrops; PHA, polyhydramnios; EM, electron microscopy; PE, pleural effusion; IHC, immunohistochemistry.
aGestational age for all cases except the current case;
bRecognized as non-neoplastic.
Figure & Data
References
Citations
- Congenital Peribronchial Myofibroblastic Tumor: Clinical Features, Pathology, and Surgical Considerations
Kavya Rajesh, Drew Bolster, Mariam Naqvi, Sonya Fabricant, Raghavendra Pillappa, Andrew Brownlee, Carlos Pelayo, Eugene Kim, David Bliss, Eveline Shue
Pediatric Blood & Cancer.2026;[Epub] CrossRef - Congenital Peribronchial Myofibroblastic Tumors Harbor a Recurrent EGFR Kinase Domain Duplication
Sheren Younes, Carlos J. Suarez, Jennifer Pogoriler, Tricia Bhatti, Archana Shenoy, Raya Saab, Lea F. Surrey, Serena Y. Tan
Modern Pathology.2025; 38(2): 100661. CrossRef - EGFR‐KDD Myofibroblastic Neoplasm or Congenital Peribronchial Myofibroblastic Tumor (CPMT)? Report of a Congenital Myofibroblastic Neoplasm With Unusual Histologic Features
Emma Rullo, Sabina Barresi, Sabrina Rossi, Sara Patrizi, Evelina Miele, Marta Barisella, Michela Casanova, Andrea Ferrari, Stefano Chiaravalli, Gloria Pelizzo, Rita Alaggio
Genes, Chromosomes and Cancer.2025;[Epub] CrossRef - Congenital peribronchial myofibroblastic tumor (CPMT): a case report with long term follow-up and next-generation sequencing (NGS)
Ping Zhou, Shuang Li, Weiya Wang, Yuan Tang, Lili Jiang
BMC Pediatrics.2023;[Epub] CrossRef - Neonatal congenital lung tumors — the importance of mid-second-trimester ultrasound as a diagnostic clue
Stephan L. Waelti, Laurent Garel, Dorothée Dal Soglio, Françoise Rypens, Michael Messerli, Josée Dubois
Pediatric Radiology.2017; 47(13): 1766. CrossRef - Congenital peribronchial myofibroblastic tumor: Case report and review of literature
Jolanta Jedrzkiewicz, Eric Scaife, Bo Hong, Sarah South, Mouied Alashari
Journal of Pediatric Surgery Case Reports.2015; 3(4): 154. CrossRef - Perinatal Thoracic Mass Lesions: Pre- and Postnatal Imaging
Evan J. Zucker, Monica Epelman, Beverley Newman
Seminars in Ultrasound, CT and MRI.2015; 36(6): 501. CrossRef - Prenatal imaging and immunohistochemical analysis of congenital peribronchial myofibroblastic tumor
Y.‐A. Tu, W.‐C. Lin, H.‐J. Chen, J.‐C. Shih
Ultrasound in Obstetrics & Gynecology.2015; 46(2): 247. CrossRef - A Congenital Peribronchial Myofibroblastic Tumor Detected in a Premature Infant at 28 Weeks but That Resolved in the Late Stage of Pregnancy
Bo Xia, Gang Yu, Chun Hong, Lei Zhang, Jing Tang, Cuifen Liu
Medicine.2015; 94(42): e1842. CrossRef - Congenital peribronchial myofibroblastic tumor
Yuka Hotokebuchi, Kenichi Kohashi, Satoshi Toyoshima, Naoko Matsumoto, Toshinori Nakashima, Yoshinao Oda
Pathology International.2014; 64(4): 189. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
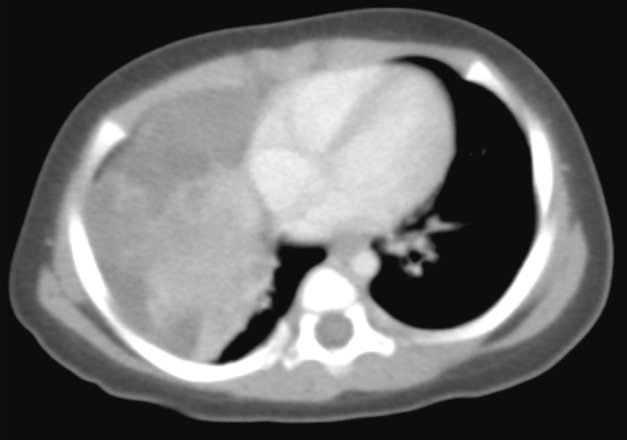
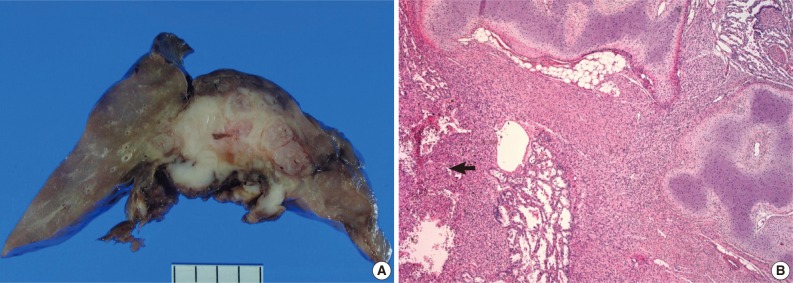
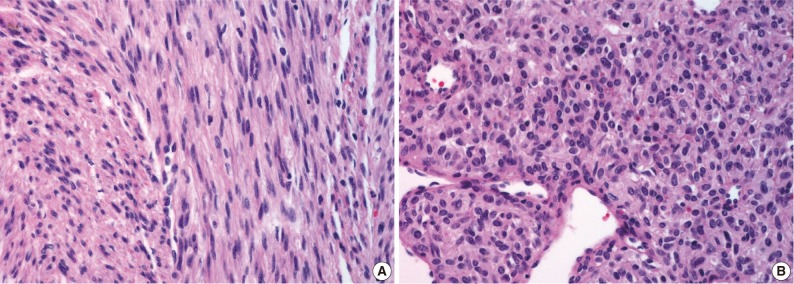
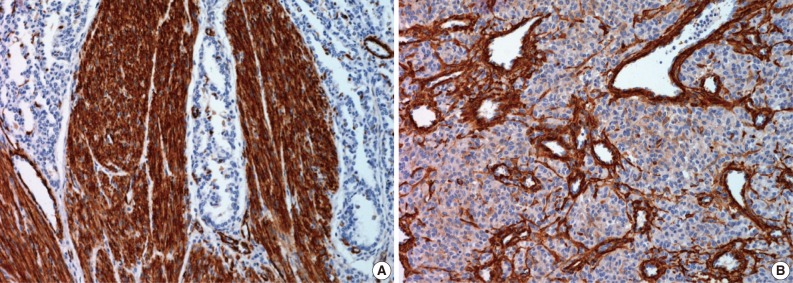
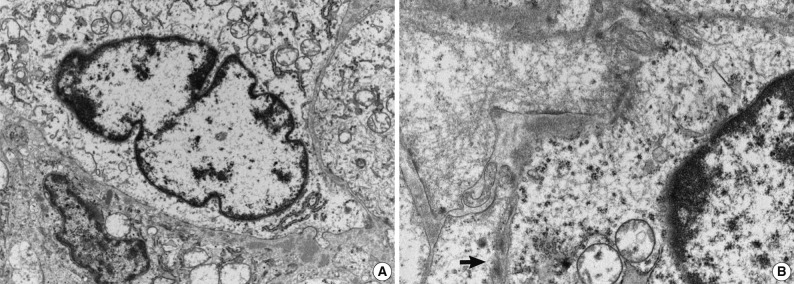





Fig. 1
Fig. 2
Fig. 3
Fig. 4
Fig. 5
| Reference | Sex/Age |
Outcome | Complication | Original diagnosis | Adjunct study | Cartilage | Necrosis | Round cell | Mitosis |
|---|---|---|---|---|---|---|---|---|---|
| Jones [9] | F/28 wk | Died at 1 hr | RD | Hamartoma | - | + | NS | NS | NS |
| Robb [3] | M/NS | Well at 10 mo | None | Fibrosarcoma | - | NS | NS | + | Not numerous |
| Guccion and Rosen [5] | M/NS | Died shortly after birth | NS | Leiomyosarcoma | - | NS | NS | NS | 15/10 hpf |
| Haller et al. [8] | F/40 wk | Died at 8 hr | H, RD | Mesenchymal tumor | - | NS | NS | + | None |
| Warren et al. [7] | F/30-33 wk | Intraoperative death | PHA, RD | Mesenchymal malformation | EM | Entrapped |
NS | NS | 1-5/hpf |
| Jimenez et al. [4] | M/36 wk | Well at 34 mo | H, PHA, RD | Leiomyosarcoma | EM | NS | NS | NS | Occasional |
| Pettinato et al. [2] | M/NS | Well at 6 yr | PE, RD | Fibrosarcoma | IHC, EM | Entrapped | + | + | 8-12/10 hpf |
| M/NS | Well at 3 mo | PE, RD | |||||||
| Khong and Keeling [6] | M/27 wk | Termination | H, PHA | Mesenchymal | - | + | NS | NS | Frequent |
| McGinnis et al. [1] | M/33 wk | Intraoperative death | H, PHA, RD | CPMT | IHC, EM, flow cytometry | + | - | NS | 1/10 hpf |
| Alobeid et al. [17] | F/35 wk | Well at 12 mo | RD | CPMT | IHC, EM, karyotyping | Entrapped | + | NS | 0-3/10 hpf |
| Kuhnen et al. [10] | NS/NS | Well at 8 mo | None | Fibrosarcoma | IHC | + | - | NS | 1/10 hpf |
| Reiss et al. [13] | M/24 wk | Termination | NS | CPMT | IHC | - | + | NS | 0-4/10 hpf |
| Horikoshi et al. [12] | M/30 wk | Intrauterine death | H, PE | CPMT | IHC | - | + | NS | NS |
| Dishop [14] | M/28 wk | Died 24 hr | H, RD | CPMT | IHC | + | - | NS | Occasional |
| de Noronha et al. [15] | M/24 wk | Intrauterine death | H, PHA | CPMT | IHC, EM | + | NS | NS | Frequent |
| Huppmann et al. [16] | M/23 wk, 6 wk postnatal | Well at 6.5 yr | H, RD | CPMT | IHC, karyotyping | + | - | NS | Brisk/occasional |
| Current case | F/1 mo | Well at 2 yr | None | CPMT | IHC, EM | + | + | + | 8/10 hpf |
CPMT, congenital peribronchial myofibroblastic tumor; F, female; RD, respiratory distress; NS, not specified; M, male; hpf, high power fields; H, hydrops; PHA, polyhydramnios; EM, electron microscopy; PE, pleural effusion; IHC, immunohistochemistry. Gestational age for all cases except the current case; Recognized as non-neoplastic.

