 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 54(1); 2020 > Article
-
Review
Molecular characteristics of meningiomas -
Young Suk Lee
 , Youn Soo Lee
, Youn Soo Lee -
Journal of Pathology and Translational Medicine 2020;54(1):45-63.
DOI: https://doi.org/10.4132/jptm.2019.11.05
Published online: January 15, 2020
Department of Hospital Pathology, College of Medicine, The Catholic University of Korea, Seoul, Korea
- Corresponding Author: Youn Soo Lee, MD, PhD, Department of Hospital Pathology, College of Medicine, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu, Seoul 06591, Korea Tel: +82-2-2258-1626, Fax: +82-2-2258-1628, E-mail: lys9908@catholic.ac.kr
• Received: August 30, 2019 • Revised: October 22, 2019 • Accepted: November 5, 2019
© 2020 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Abstract
- HISTOPATHOLOGICAL CLASSIFICATION OF MENINGIOMAS
- GENETIC ALTERATIONS IN MENINGIOMAS
- CYTOGENETIC PROGRESSION OF MENINGIOMAS
- GERMLINE ALTERATIONS AND FAMILIAL SYNDROMES PREDISPOSING MENINGIOMAS
- SOMATIC MUTATIONS IN MENINGIOMAS
- EPIGENETIC ALTERATIONS IN MENINGIOMAS
- ABERRANT SIGNALING PATHWAYS IN MENINGIOMAS
- CLINICOPATHOLOGICAL FEATURES ASSOCIATED WITH GENETIC ALTERATION IN MENINGIOMAS
- CONCLUSION
- NOTES
- REFERENCES
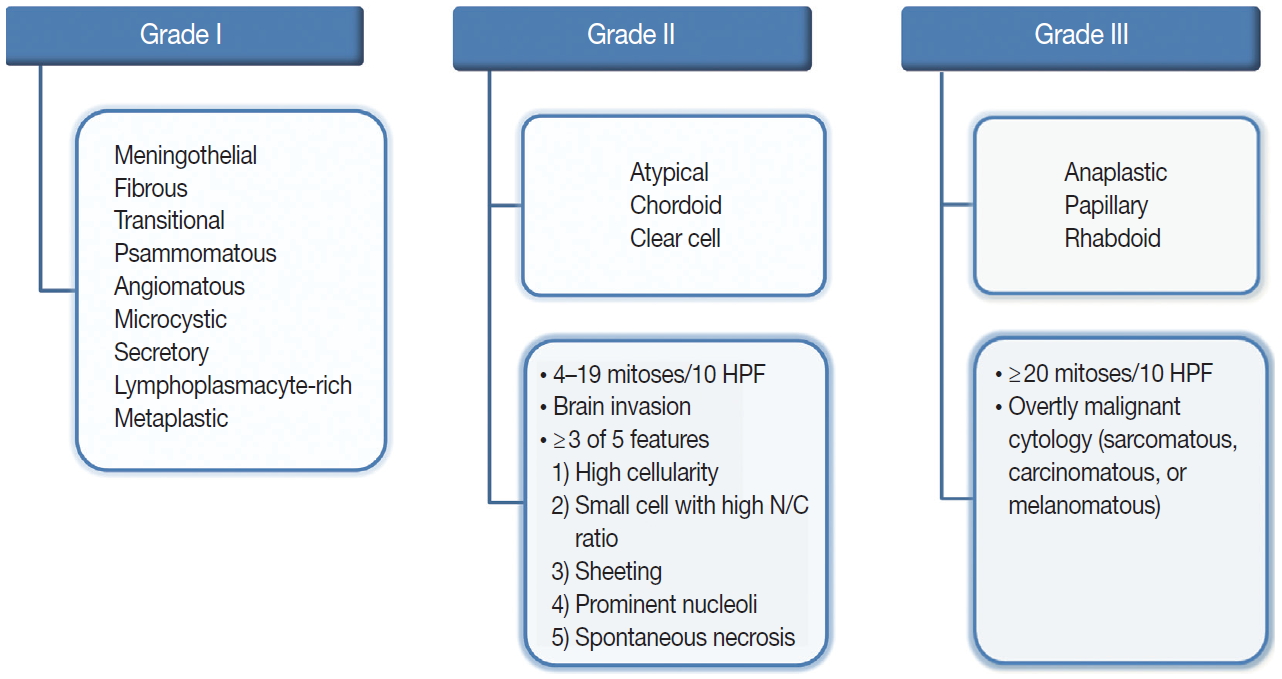
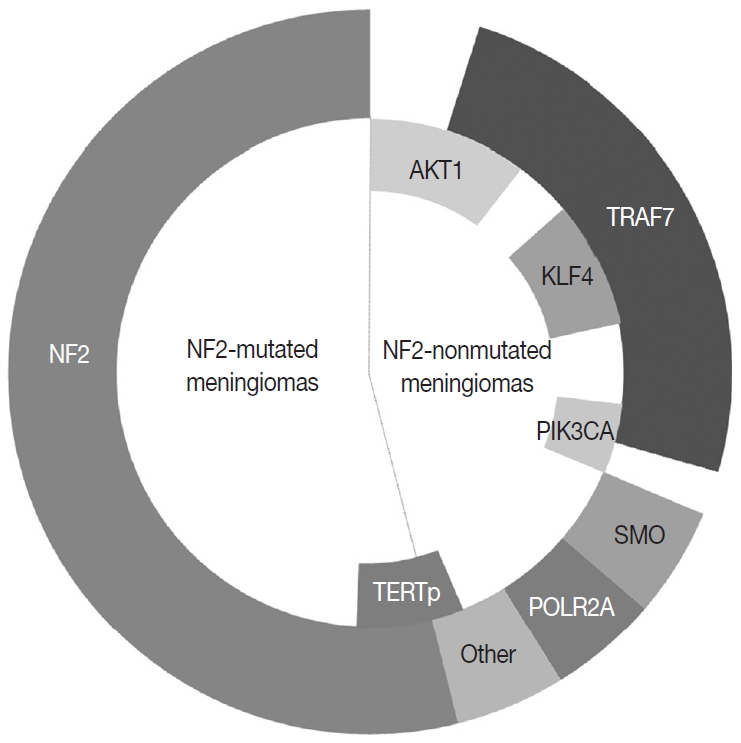
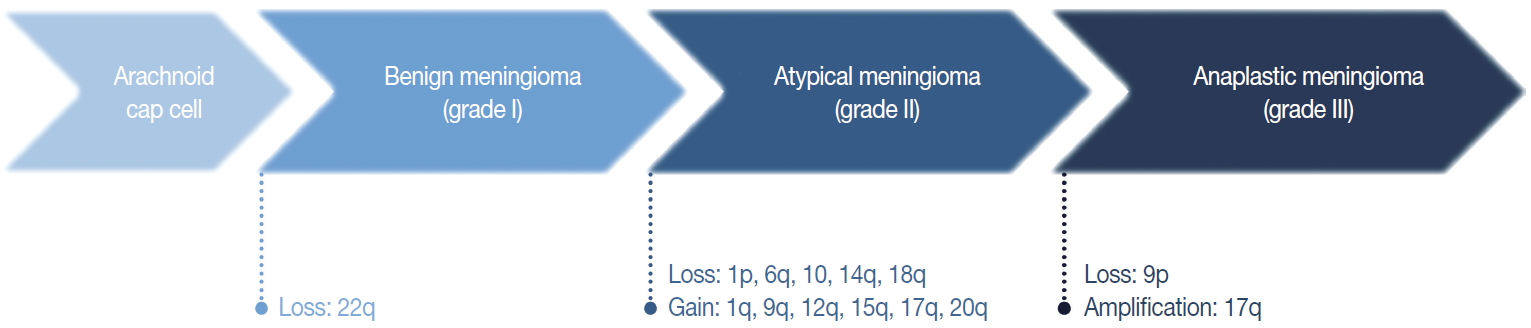
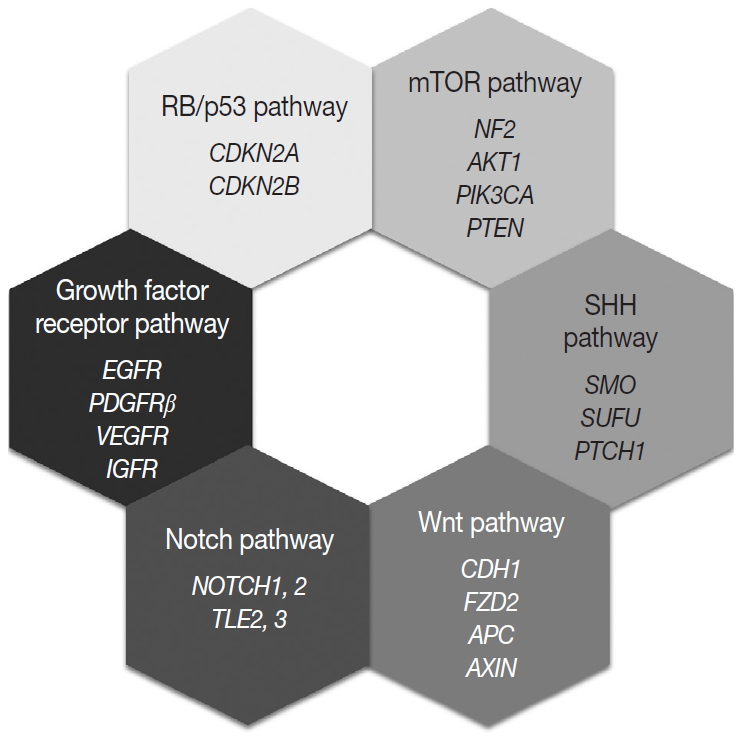
Figure & Data
References
Citations
Citations to this article as recorded by 
- Multi-Institutional Modified Delphi For Genomics in Expert Consensus Survey of Genomic Testing for Anterior Skull Base Malignancies
Anirudh Saraswathula, Shreya Sriram, Corinna Levine, Nyall R. London, Shirley Y. Su, Mathew Geltzeiler, Sanjeet V. Rangarajan, Ian Witterick, Brian Thorp, Kathleen Kelly Gallagher, Kenneth Byrd, Ricardo Carrau, Waleed Abuzeid, Eric Wang, Carl Snyderman, E
Journal of Neurological Surgery Part B: Skull Base.2026; 87(01): 014. CrossRef - Long-Term Prognostic Impact of the Dural Tail in the Local Control of Intracranial Meningiomas
Franco Rubino, Romulo A. de Almeida, Komal Shah, Shaan M. Raza, Franco DeMonte
Journal of Neurological Surgery Part B: Skull Base.2026;[Epub] CrossRef - Multidisciplinary management of meningiomas in the era of precision oncology
Majid Assadi, Malik E. Juweid, Philipp Lohmann, Giuseppe Minniti, Felix Sahm, Jana Ivanidze, Philipp Karschnia, Alexander Landry, Felix M. Mottaghy, Gelareh Zadeh
Nature Reviews Clinical Oncology.2026; 23(7): 539. CrossRef - From imaging to molecular pathways: a comprehensive narrative review of the distinctive landscape of secretory meningioma
Erfan Shahabinejad, Mahsa Kamali, Amirreza Shakoeizadeh, Zeinab Falakian, Aynaz Foroughi Eghbal, Seyyed Mohammad Alipour, Maria Krywyj, Jason P. Sheehan, Qais Alrashidi, Hamid Borghei-Razavi
International Journal of Neuroscience.2026; : 1. CrossRef - Recurrence of Resected Skull Base Meningiomas during Long-term Follow-up: Incidence and Predisposing Factors
Joshua Ian Macarthur, Cathal John Hannan, Callum Howard, Jane Halliday, Omar Nathan Pathmanaban, Charlotte Hammerbeck-Ward, Scott A. Rutherford, Andrew T. King
Journal of Neurological Surgery Part B: Skull Base.2025; 86(03): 245. CrossRef - Role of H3K27me3 and Ki-67 Labeling Index in Assessing the Biological Behavior of Meningiomas
Shalaka Deshpande, Bhavna Nayal, Rajesh Nair, Deepak Nayak, Padmapriya J, Geetha V
World Neurosurgery.2025; 194: 123514. CrossRef - Context aware machine learning techniques for brain tumor classification and detection – A review
Usman Amjad, Asif Raza, Muhammad Fahad, Doaa Farid, Adnan Akhunzada, Muhammad Abubakar, Hira Beenish
Heliyon.2025; 11(2): e41835. CrossRef - Case report: Clonal evolution analysis of a rare case of meningioma lung metastases identifies actionable alterations in matched longitudinal tumour samples
Nicola Cosgrove, Orla M. Fitzpatrick, Liam Grogan, Bryan T. Hennessy, Simon J. Furney, Sinead Toomey
Frontiers in Oncology.2025;[Epub] CrossRef - Post-operative Hemorrhage After Tumor Removal of Multiple Meningiomas
Bob Irfan Syahputra, Akhmad Imron, Dhany Febriantara, Helza Efriani
International Journal of Recent Surgical and Medical Sciences.2025; 11: e005. CrossRef - Diarylpentanoid, a curcumin analog, inhibits malignant meningioma growth in both in vitro and in vivo models
Anna Terasawa, Kazuhiro Shimazu, Hiroshi Nanjo, Masatomo Miura, Hiroyuki Shibata
World Journal of Experimental Medicine.2025;[Epub] CrossRef - Tumour-associated macrophages in human meningiomas
Rahmina Meta, Henrik Sahlin Pettersen, Sofie Eline Tollefsen, Borgny Ytterhus, Øyvind Olav Salvesen, Wenche Sjursen, Sverre Helge Torp, Jianhong Zhou
PLOS One.2025; 20(5): e0319960. CrossRef - Binary Classification of Meningioma Grades Using CNN and VGG16 + XGBoost Deep Learning Models
L. Priya, D. Saraswathi, M. Bhuvaneshwari, K. Dhanya, K. Krishna Kousalya, Deepali
Applied Computational Intelligence and Soft Computing.2025;[Epub] CrossRef - Unusual adrenal metastasis of anaplastic meningioma: A case report
M. Inouss, C. Rhoul, A. Kharkhach, T. Bouhout, B. Serji
International Journal of Surgery Case Reports.2025;[Epub] CrossRef - TERT promoter mutations in meningiomas: associations with clinicopathological features and insights into spatial and temporal heterogeneity in a 165-case cohort
Ahmet Boduroğlu, Mualla Özcan, Güzide Ayşe Ocak
Pathology - Research and Practice.2025; 274: 156191. CrossRef - The Gut–Brain Axis in Brain Tumors: Insights into Tumor Development, Progression, and Therapy
Sarah Adriana Scuderi, Alessio Ardizzone, Elsa Calcaterra, Nicoletta Palermo, Fabiola De Luca, Antonio Catalfamo, Emanuela Esposito, Anna Paola Capra
Biomedicines.2025; 13(9): 2172. CrossRef - The Natural History and Treatment of Meningiomas: An Update
Arsene Daniel Nyalundja, Fabrice Mugisha, Claire Karekezi
Seminars in Neurology.2024; 44(01): 001. CrossRef - Epidemiology, Genetics, and DNA Methylation Grouping of Hyperostotic Meningiomas
Gray Umbach, Edwina B. Tran, Charlotte D. Eaton, Abrar Choudhury, Ramin Morshed, Javier E. Villanueva-Meyer, Philip V. Theodosopoulos, Stephen T. Magill, Michael W. McDermott, David R. Raleigh, Ezequiel Goldschmidt
Operative Neurosurgery.2024; 26(6): 662. CrossRef - The Evolving Classification of Meningiomas: Integration of Molecular Discoveries to Inform Patient Care
S. Joy Trybula, Mark W. Youngblood, Constantine L. Karras, Nikhil K. Murthy, Amy B. Heimberger, Rimas V. Lukas, Sean Sachdev, John A. Kalapurakal, James P. Chandler, Daniel J. Brat, Craig M. Horbinski, Stephen T. Magill
Cancers.2024; 16(9): 1753. CrossRef - Minimally Invasive Approaches in the Surgical Treatment of Intracranial Meningiomas: An Analysis of 54 Cases
Guenther C. Feigl, Daniel Staribacher, Gavin Britz, Dzmitry Kuzmin
Brain Tumor Research and Treatment.2024; 12(2): 93. CrossRef - Related mechanisms, current treatments, and new perspectives in meningioma
Gizem Inetas‐Yengin, Omer Faruk Bayrak
Genes, Chromosomes and Cancer.2024;[Epub] CrossRef - Clinical application of intraoperative ultrasound superb microvascular imaging in brain tumors resections: contributing to the achievement of total tumoral resection
Siman Cai, Hao Xing, Yuekun Wang, Yu Wang, Wenbin Ma, Yuxin Jiang, Jianchu Li, Hongyan Wang
BMC Medical Imaging.2024;[Epub] CrossRef - WHO CNS 5 and meningiomas: What’s new?
Indranil Chakrabarti, Sujaya Mazumder
IP Archives of Cytology and Histopathology Research.2024; 9(2): 67. CrossRef - Molecular Developments in Parasellar Tumors and Potential Therapeutic Implications
Paraskevi Xekouki, Vasiliki Venetsanaki, Georgios Kyriakopoulos, Krystallenia Alexandraki, Anna Angelousi, Gregory Kaltsas
Endocrine Reviews.2024; 45(6): 880. CrossRef - The Impact of Molecular and Genetic Analysis on the Treatment of Patients with Atypical Meningiomas
Janez Ravnik, Hojka Rowbottom
Diagnostics.2024; 14(16): 1782. CrossRef - Differential Expression of Proteins and Genes at the Tumor‐Brain Interface in Invasive Meningioma
Kornwika Senglek, Chinachote Teerapakpinyo, Nutchawan Jittapiromsak, Pakrit Jittapiromsak, Irin Lertparinyaphorn, Paul Scott Thorner, Shanop Shuangshoti
Genes, Chromosomes and Cancer.2024;[Epub] CrossRef - Protein expression of CD44 in patients with meningioma tumors: association with clinicopathological parameters and survival
Trupti Trivedi, Neha Bhalala, Kirti Dialani, Priti Trivedi
Journal of the Egyptian National Cancer Institute.2024;[Epub] CrossRef - DNA methylation profiling of meningiomas highlights clinically distinct molecular subgroups
Jyotsna Singh, Ravi Sharma, Nidhi Shukla, Priya Narwal, Amit Katiyar, Swati Mahajan, Saumya Sahu, Ajay Garg, Mehar C. Sharma, Ashish Suri, Chitra sarkar, Vaishali Suri
Journal of Neuro-Oncology.2023; 161(2): 339. CrossRef - Spinal meningiomas, from biology to management - A literature review
Nicolas Serratrice, Imène Lameche, Christian Attieh, Moussa A Chalah, Joe Faddoul, Bilal Tarabay, Rabih Bou-Nassif, Youssef Ali, Joseph G Mattar, François Nataf, Samar S Ayache, Georges N Abi Lahoud
Frontiers in Oncology.2023;[Epub] CrossRef - Somatic mutation landscape in a cohort of meningiomas that have undergone grade progression
Sarah A Cain, Bernard Pope, Stefano Mangiola, Theo Mantamadiotis, Katharine J Drummond
BMC Cancer.2023;[Epub] CrossRef - Actualización sobre el meningioma: correlación clínico-radiológica y radio-patológica
A. Navarro-Ballester, M. Aleixandre-Barrachina, S.F. Marco-Doménech
Radiología.2023; 65(5): 458. CrossRef - SMARCE1-related meningiomas: A clear example of cancer predisposing syndrome
Erika Fiorentini, Laura Giunti, Andrea Di Rita, Simone Peraio, Carla Fonte, Chiara Caporalini, Anna Maria Buccoliero, Maria Luigia Censullo, Giulia Gori, Alice Noris, Rosa Pasquariello, Roberta Battini, Rossana Pavone, Flavio Giordano, Sabrina Giglio, Ber
European Journal of Medical Genetics.2023; 66(7): 104784. CrossRef - Grade scoring system reveals distinct molecular subtypes and identifies KIF20A as a novel biomarker for predicting temozolomide treatment efficiency in gliomas
Liguo Ye, Shi’ao Tong, Yaning Wang, Yu Wang, Wenbin Ma
Journal of Cancer Research and Clinical Oncology.2023; 149(12): 9857. CrossRef - Integrated clinical genomic analysis reveals xenobiotic metabolic genes are downregulated in meningiomas of current smokers
A. Basit Khan, Rajan Patel, Malcolm F. McDonald, Eric Goethe, Collin English, Ron Gadot, Arya Shetty, Shervin Hosseingholi Nouri, Arif O. Harmanci, Akdes S. Harmanci, Tiemo J. Klisch, Akash J. Patel
Journal of Neuro-Oncology.2023; 163(2): 397. CrossRef - DNA methylation meningioma biomarkers: attributes and limitations
Zhaohui Li, Yufei Gao, Jinnan Zhang, Liang Han, Hang Zhao
Frontiers in Molecular Neuroscience.2023;[Epub] CrossRef - Meningioma: A Biography—Tumor Forever Tied to the Origins and “Soul of Neurosurgery”
Nolan J. Brown, Zach Pennington, Cathleen C. Kuo, Julian Gendreau, Sachiv Chakravarti, Rohin Singh, Dontré M. Douse, Jamie J. Van Gompel
World Neurosurgery.2023; 178: 191. CrossRef - Molecular genetic features of meningiomas
E.S. Makashova, N.V. Lasunin, M.V. Galkin, S.V. Zolotova, K.O. Karandasheva, A.V. Golanov
Burdenko's Journal of Neurosurgery.2023; 87(4): 101. CrossRef - Novel Advances in Treatment of Meningiomas: Prognostic and Therapeutic Implications
Gerardo Caruso, Rosamaria Ferrarotto, Antonello Curcio, Luisa Metro, Francesco Pasqualetti, Paola Gaviani, Valeria Barresi, Filippo Flavio Angileri, Maria Caffo
Cancers.2023; 15(18): 4521. CrossRef - Update on meningioma: Clinical-radiological and radio-pathological correlation
A. Navarro-Ballester, M. Aleixandre-Barrachina, S.F. Marco-Doménech
Radiología (English Edition).2023; 65(5): 458. CrossRef - Early Preventive Strategies and CNS Meningioma – Is This Feasible? A Comprehensive Review of the Literature
Daniel Sescu, Aminta Chansiriwongs, Katarzyna Julia Minta, Jyothi Vasudevan, Chandrasekaran Kaliaperumal
World Neurosurgery.2023; 180: 123. CrossRef - Domestic Animal Models of Central Nervous System Tumors: Focus on Meningiomas
Michele Tomanelli, Tullio Florio, Gabriela Vargas, Aldo Pagano, Paola Modesto
Life.2023; 13(12): 2284. CrossRef - Assessment of parameters of the acid-base state among patients with meningiomas and gliomas in the postoperative period
E. S. Orlova, I. O. Ishchenko, K. K. Kukanov, N. E. Voinov, A. P. Gerasimov, N. E. Ivanova
Russian Neurosurgical Journal named after Professor A. L. Polenov.2023; 15(2): 21. CrossRef - The integrated multiomic diagnosis of sporadic meningiomas: a review of its clinical implications
Stephanie M. Robert, Shaurey Vetsa, Arushii Nadar, Sagar Vasandani, Mark W. Youngblood, Evan Gorelick, Lan Jin, Neelan Marianayagam, E Zeynep Erson-Omay, Murat Günel, Jennifer Moliterno
Journal of Neuro-Oncology.2022; 156(2): 205. CrossRef - Clinical presentation, diagnostic findings and outcome of dogs undergoing surgical resection for intracranial meningioma: 101 dogs
Alexander K. Forward, Holger Andreas Volk, Giunio Bruto Cherubini, Tom Harcourt-Brown, Ioannis N. Plessas, Laurent Garosi, Steven De Decker
BMC Veterinary Research.2022;[Epub] CrossRef - Sphenoid wing meningiomas: peritumoral brain edema as a prognostic factor in surgical outcome
Abdalrahman Nassar, Volodymyr Smolanka, Andriy Smolanka, Dipak Chaulagain, Oleg Devinyak
Neurosurgical Review.2022; 45(4): 2951. CrossRef - Potential Molecular Mechanisms of Recurrent and Progressive Meningiomas: A Review of the Latest Literature
Wenjie Peng, Pei Wu, Minghao Yuan, Bo Yuan, Lian Zhu, Jiesong Zhou, Qian Li
Frontiers in Oncology.2022;[Epub] CrossRef - Case Report: Upper Thoracic Purely Extradural Spinal Meningioma With Nerve Root Attachment: A Case Report and Literature Review
Zhao-Lin Wang, Jian-Hui Mou, Dong Sun, Peng Liu
Frontiers in Surgery.2022;[Epub] CrossRef - Molecular diagnosis and treatment of meningiomas: an expert consensus (2022)
Jiaojiao Deng, Lingyang Hua, Liuguan Bian, Hong Chen, Ligang Chen, Hongwei Cheng, Changwu Dou, Dangmurenjiapu Geng, Tao Hong, Hongming Ji, Yugang Jiang, Qing Lan, Gang Li, Zhixiong Liu, Songtao Qi, Yan Qu, Songsheng Shi, Xiaochuan Sun, Haijun Wang, Yongpi
Chinese Medical Journal.2022; 135(16): 1894. CrossRef - Оновлена інформація про менінгіоми крила клиноподібної кістки
Abdalrahman Nassar, Volodymyr Smolanka
INTERNATIONAL NEUROLOGICAL JOURNAL.2022; 18(1): 43. CrossRef - The Prognostic Value of Methylation Signatures and NF2 Mutations in Atypical Meningiomas
Rahmina Meta, Henning B. Boldt, Bjarne W. Kristensen, Felix Sahm, Wenche Sjursen, Sverre H. Torp
Cancers.2021; 13(6): 1262. CrossRef - Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis
Suha Bachir, Sanjit Shah, Scott Shapiro, Abigail Koehler, Abdelkader Mahammedi, Ravi N. Samy, Mario Zuccarello, Elizabeth Schorry, Soma Sengupta
International Journal of Molecular Sciences.2021; 22(2): 690. CrossRef - Meningioma: A Review of Epidemiology, Pathology, Diagnosis, Treatment, and Future Directions
Christian Ogasawara, Brandon D. Philbrick, D. Cory Adamson
Biomedicines.2021; 9(3): 319. CrossRef - The substantial loss of H3K27me3 can stratify risk in grade 2, but not in grade 3 meningioma
Minsun Jung, Seong-Ik Kim, Ka Young Lim, Jeongmo Bae, Chul-Kee Park, Seung Hong Choi, Sung-Hye Park, Jae-Kyung Won
Human Pathology.2021; 115: 96. CrossRef - Papillary Meningioma: Case Presentation with Emphasis on Surgical and Medical Therapy of a Rare Variant of Meningioma
Gerardo Cazzato, Valeria Internò, Antonietta Cimmino, Raffaella Messina, Marco Tucci, Teresa Lettini, Leonardo Resta, Giuseppe Ingravallo
Diseases.2021; 9(3): 63. CrossRef - An Overview of Managements in Meningiomas
Lianhua Zhao, Wei Zhao, Yanwei Hou, Cuixia Wen, Jing Wang, Pei Wu, Zaiyu Guo
Frontiers in Oncology.2020;[Epub] CrossRef - Multi-Omics Analysis in Initiation and Progression of Meningiomas: From Pathogenesis to Diagnosis
Jiachen Liu, Congcong Xia, Gaiqing Wang
Frontiers in Oncology.2020;[Epub] CrossRef - Molecular Mechanism and Approach in Progression of Meningioma
Zhiwei Shao, Lihong Liu, Yanghao Zheng, Sheng Tu, Yuanbo Pan, Sheng Yan, Qichun Wei, Anwen Shao, Jianmin Zhang
Frontiers in Oncology.2020;[Epub] CrossRef - Multiple meningiomas: does quantity matter? a population-based survival analysis with underlined age and sex differences
Andres Ramos-Fresnedo, Ricardo A. Domingo, Tito Vivas-Buitrago, Larry Lundy, Daniel M. Trifiletti, Mark E. Jentoft, Amit B. Desai, Alfredo Quiñones-Hinojosa
Journal of Neuro-Oncology.2020; 149(3): 413. CrossRef - Meningioma: A Review of Clinicopathological and Molecular Aspects
Kristin Huntoon, Angus Martin Shaw Toland, Sonika Dahiya
Frontiers in Oncology.2020;[Epub] CrossRef - Neues zur Einteilung und Therapie von Meningeomen
Corinna Seliger, Wolfgang Wick
Neurologie up2date.2020; 3(04): 343. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-

Molecular characteristics of meningiomas




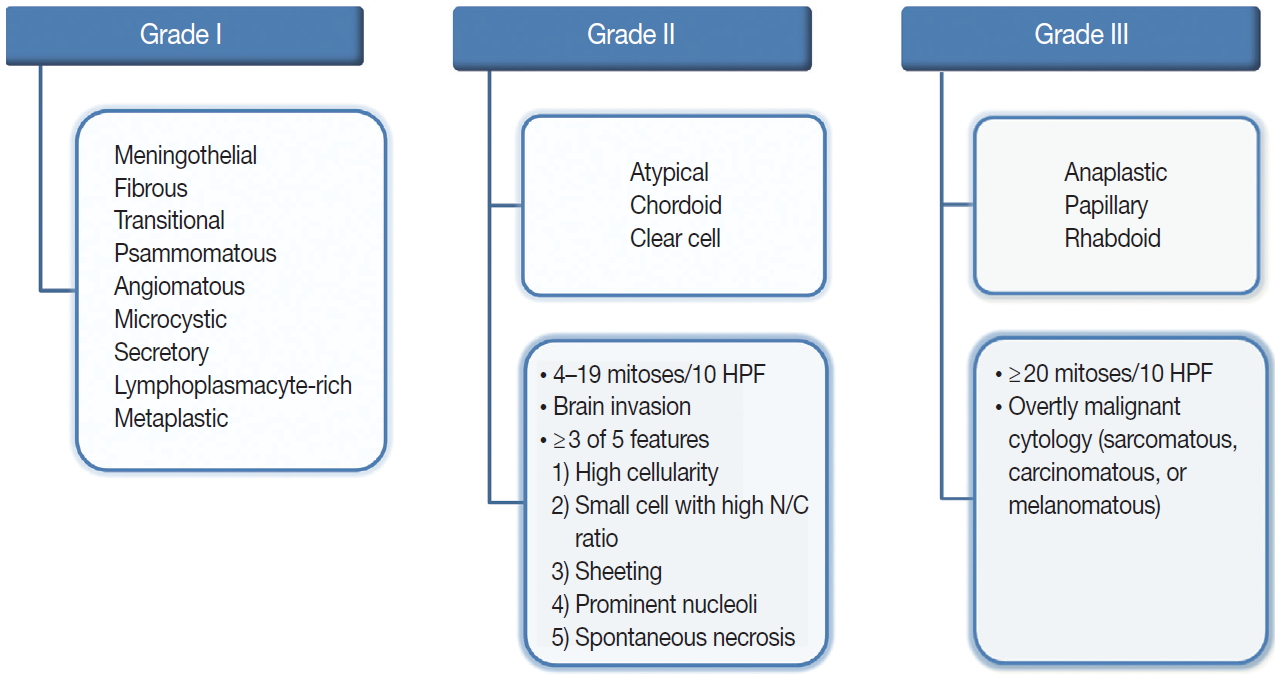
Fig. 1. Overview of the 2016 World Health Organization classification system for grading meningiomas. HPF, high-power fields; N/C, nuclear/cytoplasmic.
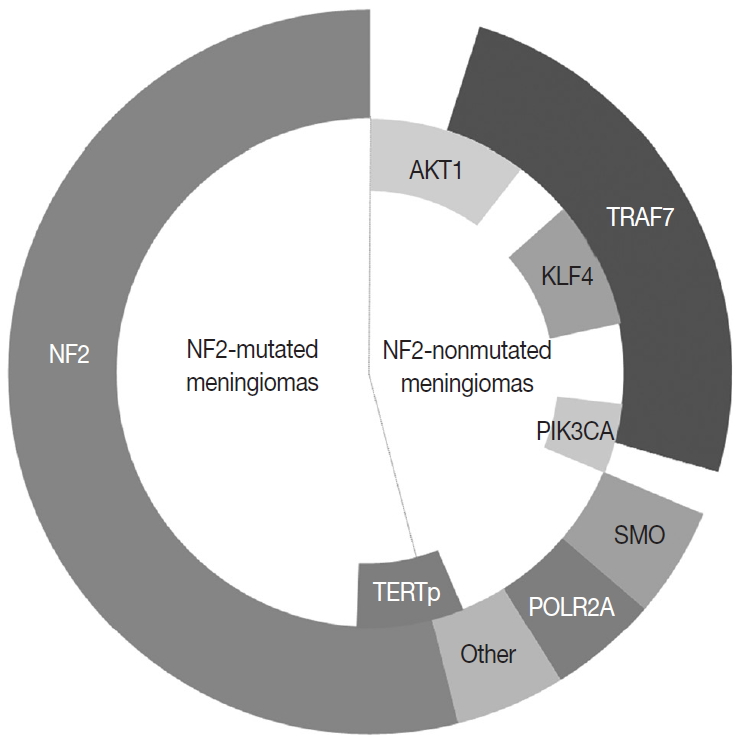
Fig. 2. The most common genetic alterations found in meningiomas.
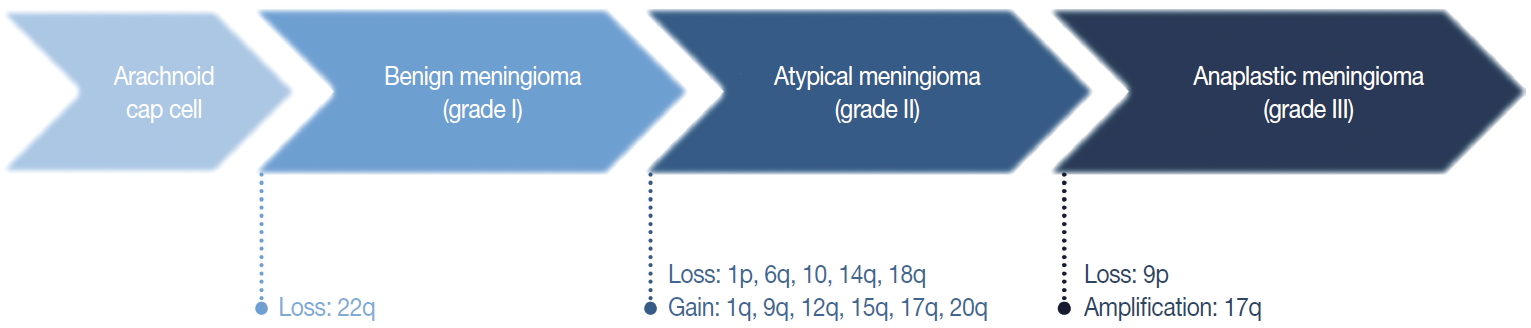
Fig. 3. Cytogenetic changes during initiation and progression of meningiomas.
Fig. 4. Molecular signaling pathways associated with meningiomas.
Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Molecular characteristics of meningiomas
| Familial syndrome | Affected gene | Chromosome locus |
|---|---|---|
| Neurofibromatosis type 2 | NF2 | 22q12 |
| Familial schwannomatosis | SMARCB1 | 22q11.23 |
| Multiple spinal meningiomas | SMARCE1 | 17q21.2 |
| BAP1 tumor predisposition syndrome | BAP1 | 3p21.1 |
| Gorlin syndrome (nevoid basal cell carcinoma syndrome) | PTCH1 | 9q22.3 |
| SUFU | 10q24.32 | |
| Familial multiple meningiomas | SUFU | 10q24.32 |
| Rubinstein-Taybi syndrome | CREBBP | 16p13.3 |
| von Hippel-Lindau syndrome | VHL | 3p25-26 |
| Cowden disease | PTEN | 10q23.31 |
| Li-Fraumeni syndrome | TP53/CHEK2 | 17p13.1/22q12.1 |
| Gardner syndrome | APC | 5q21-22 |
| Multiple endocrine neoplasia type 1 | MEN | 11q13 |
| Werner syndrome | LMNA | 1q21.1 |
| Gene | Full name | Locus | Product | Function |
|---|---|---|---|---|
| NF2 | Neurofibromin 2 | 22q12.2 | Merlin | Tumor suppressor |
| Maintain cell shape by linkage cell membrane proteins to cytoskeleton | ||||
| TRAF7 | TNF receptor-associated factor 7 | 16p13.3 | TNF receptor-associated factor 7 | E3 ubiquitin ligase |
| Interaction with MAPK pathway | ||||
| KLF4 | Kruppel-like factor 4 | 9p31 | Kruppel-like factor 4 | Transcription factor |
| Induction of pluripotency | ||||
| AKT1 | v-Akt murine thymoma viral oncogene homolog 1 | 14q32.33 | AKT1 kinase (serine/threonine protein kinase) | Oncogene |
| Regulate cell growth and division, control apoptosis | ||||
| SMO | Smoothened, frizzled class receptor | 7p32.1 | Smoothened, G protein-coupled receptor | Activation of hedgehog pathway |
| Cell growth, proliferation | ||||
| PIK3CA | Phosphadidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha | 3q26.32 | Catalytic subunit of kinase, PI3K | Activation of PI3K/AKT pathway |
| POLR2A | RNA polymerase II subunit A | 17p13.1 | RNA polymerase II subunit A | Formation of preinitiation complex for transcription |
| FAK | Focal adhesion kinase | 8q24.3 | Protein tyrosine kinase2 | Cytoplasmic tyrosine kinase |
| Cellular proliferation and motility | ||||
| BAP1 | BRCA1-associated protein 1 | 3p21.1 | Ubiquitin carboxyl-terminal hydrolase 1 | Deubiquitinase, tumor suppressor |
| Control cell growth, division and cell death | ||||
| SMARCB1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 | 22q11.23 | Subunit of SWI/SNF complex | Tumor suppressor |
| Regulate gene activity by chromatin remodeling | ||||
| SMARCE1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily e, member 1 | 17q21.2 | Subunit of SWI/SNF complex | Tumor suppressor |
| Regulate gene activity by chromatin remodeling | ||||
| BRAF V600E | B-Raf proto-oncogene | 7q34 | Serine/threonine kinase | Oncogene |
| Involved in MAPK pathway | ||||
| NOTCH2 | Notch receptor 2 | 1p12 | Notch2 (notch receptor family) | Transmembrane receptor |
| Involved in Notch signaling pathway | ||||
| CHEK2 | Checkpoint kinase2 | 22q12.1 | Checkpoint kinase 2 (CHK2) | Tumor suppressor |
| Regulate cell division | ||||
| PTEN | Phosphatase and tensin homolog | 10q23.31 | Phosphatidylinositol-3,4,5-triphosphate 3-phosphatase | Tumor suppressor |
| Negative regulation of mTOR pathway | ||||
| CDKN2A | Cyclin-dependent kinase inhibitor 2A | 9p21.3 | p16(INK4A) | Tumor suppressor |
| p14(ARF) | Cell cycle progression (G1/S phase) | |||
| CDKN2B | Cyclin-dependent kinase inhibitor 2B | 9p21.3 | p15(INK4B) | Tumor suppressor |
| Cell cycle progression (G1/S phase) | ||||
| DMD | Dystrophin | Xp21.1 | Dystrophin | Cytoskeletal construction, cell proliferation and motility |
| TERTp | Telomerase reverse transcriptase | 5p15.33 | Telomerase reverse transcriptase | Maintain telomere end |
| Affected gene | Product | Physiologic function | Possible role in meningioma |
|---|---|---|---|
| Focal DNA methylation | |||
| TIMP3 | Metalloproteinase inhibitor 3 | Inhibit MMP9 activity, tumor suppressor | Higher grade |
| TP73 | p73 | Cell cycle and growth regulation, tumor suppressor | Tumorigenesis |
| Malignant transformation | |||
| MEG3 | Noncoding RNA | Cell cycle, p53 activation, tumor suppressor | Tumorigenesis |
| Higher grade | |||
| GSTP1 | Glutathione S transferase 1 | Carcinogen detoxification, tumor suppressor | Higher grade |
| HOXA5, 6, 9, 11 | Homeobox A gene cluster | Organ development and cell signaling | Tumorigenesis |
| Higher grade, progression | |||
| WNK2 | Serine-threonine kinase | Negatively regulation of EGFR signaling | Tumorigenesis |
| Higher grade, progression | |||
| MAL2 | Mal proteolipid protein2 | Potential role in apoptosis | Higher grade |
| Malignant transformation | |||
| Histone modification | |||
| H3K27 | Histone H3 | Trimethylated H3K27 (H3K27m3): gene silencing | Tumorigenesis |
| Worse outcome | |||
| HIST1HIC | Histone H1.2 | Maintenance of methylation pattern | Recurrence |
| Tumor suppressor | |||
| KDM5C | Lysine demethylase 5C | Chromatin structuring | Grade I, III |
| KDM6A | Lysine demethylase 5C | Chromatin structuring | Grade I |
| Chromatin remodeler | |||
| SMARCB1 | Subunit of SWI/SNF complex | Chromatin structuring | Higher grade |
| SMARCE1 | Subunit of SWI/SNF complex | Chromatin structuring | Higher grade |
| microRNA | |||
| miR-21 | MicroRNA | Tumorigenesis, progression | |
| miR-190a | MicroRNA | Antiapoptosis | Recurrence |
| miR-335 | MicroRNA | Proliferation, RB1 signaling | Progression |
| miR-29c-3p | MicroRNA | Proliferation | Recurrence |
| miR-219-5p | MicroRNA | Proliferation, apoptosis | Recurrence |
| miR-145 | MicroRNA | Collagen regulation | Higher grade |
| Apoptosis, migration | |||
| miR-200a | MicroRNA | E-cadherin regulation | Tumorigenesis |
| Wnt/β-catenin signaling | Multifunctional | ||
| miR-224 | MircoRNA | Progression |
| Gene | Frequency (%) | Grade | Variant | Location | Prognosis |
|---|---|---|---|---|---|
| NF2 | 40–60 | I, II, III | Fibrous | Convexity | Poor for deletion |
| Transitional | Posterior skull base | ||||
| TRAF7 | 15–25 | I > II, III | Meningothelial | Skull base | Dependent on coexisting mutation |
| Secretory | |||||
| KLF4 | 9–12 | I | Secretory | Skull base | Low risk of malignant progression |
| AKT1 | 7–12 | I | Meningothelial | Anterior skull base | Early tumor recurrence |
| SMO | 1–5 | I | Meningothelial | Medial anterior skull base | Larger tumor volume |
| Olfactory groove | Higher recurrence rate | ||||
| PIK3CA | 3–4 | I > II, III | Meningothelial | Skull base | Rarely high grade, poor for Glu545Lys and His1047Arg |
| Transitional | |||||
| POLR2A | 6 | I | Meningothelial | Skull base (tuberculum sellae) | Low risk of recurrence and malignant progression |
| TERT | 6 | I < II, III | Atypical anaplastic (secondary) | Convexity skull base | High grade, poor |
| SMARCB1 | 5 | I < II, III | Falx cerebri | High grade, poor | |
| SMARCE1 | 3–4 | II | Clear cell | Spinal | High risk of recurrence |
| High grade, poor | |||||
| BAP1 | < 1 | III | Rhabdoid | Convexity | Poor |
Table 1. Familial syndromes associated with meningiomas
Table 2. Genes associated with meningiomas
Table 3. Epigenetic changes in meningiomas
Table 4. Clinicopathological features related to genetic alteration in meningiomas

