Molecular characteristics of meningiomas
Article information

Abstract
Meningioma is the most common primary intracranial tumor in adults. The grading of meningioma is based on World Health Organization criteria, which rely on histopathological features alone. This grading system is unable to conclusively predict the clinical behavior of these tumors (i.e., recurrence or prognosis in benign or atypical grades). Advances in molecular techniques over the last decade that include genomic and epigenomic data associated with meningiomas have been used to identify genetic biomarkers that can predict tumor behavior. This review summarizes the molecular characteristics of meningioma using genetic and epigenetic biomarkers. Molecular alterations that can predict meningioma behavior may be integrated into the upcoming World Health Organization grading system.
Meningioma is the most common primary neoplasm of the central nervous system (CNS) in adults [1]. It accounts for one-third (37.1%) of all primary intracranial tumors [1,2] with an incidence rate of 8.33/100,000 person/yr in the United States [2]. Meningiomas primarily occur in the elderly population and are more frequent in the 6th to 7th decades, but are exceedingly rare in childhood (0.4%–4.1% of all pediatric neoplasms) [3]. In adults, females are affected by meningioma more frequently, with a female-to-male ratio [2] of 2.27:1, whereas in childhood it occurs equally [3,4].
The majority of meningiomas (79.8%) are intracranial tumors, and 4.2% of meningiomas have spinal locations [2]. Meningothelial arachnoid cells are the precursors of meningiomas [5] and have different origins in embryogenesis according to their location [5]. The meninges of the brain convexity and skull base are developed from the neural crest and mesodermal structure, respectively [5,6]. This difference affects not only the histologic features but also the recurrent somatic mutations [5,6].
Older age and genetic susceptibility are known risk factors for meningiomas [7], and one important established cause is ionizing radiation [7,8]. As well as with high-dose radiation, therapeutic exposure to low-dose radiation (1–2 Gy), especially in childhood, has been linked to higher meningioma risk [9]. Radiation-induced meningiomas tend to present with multiple tumors and have aggressive clinicopathological features [8,9]. Other possible risk factors may include head trauma, gonadal steroid hormones, smoking, and some viral infections [7,10-13]. Several findings, such as a higher incidence in females and the expression of progesterone receptor in 76% of benign meningiomas, suggest an endocrine influence on the pathogenesis of meningiomas [13,14].
According to the World Health Organization (WHO) classification, most meningiomas (80%) are grade I, which have benign histology and indolent behavior [15]. Whereas the remaining tumors (20%) are a higher grade (grade II and III) with atypical to malignant histology and demonstrate more aggressive course [2,6].
To date, surgery is the mainstay of treatment and 70%–80% of meningiomas can be cured by surgical resection [6]. The most reliable prognostic factors of meningiomas are histologic grade (WHO grade) and the extent of resection (Simpson grade) [6,16,17]. After complete resection, the 5-year recurrence rates of WHO grade I, II, and III tumors are 5%–10%, 50%, and 80%, respectively [18].
Meningiomas in surgically challenging locations such as the skull base, meningiomas with higher histologic grade and aggressive behavior, and meningiomas that recur despite radical resection, all require another treatment strategy [6]. Radiation therapy has been used as an adjuvant modality, but systemic therapy has demonstrated limited benefits [6].
In the last decade, our understanding of the molecular profile of meningiomas has improved. Identification of genetic and epigenetic alterations in meningiomas enable the clarification of their biologic behavior and prognostic stratification. Comprehensive information on molecular features provides the opportunity of refining the classification of meningiomas that can predict tumor behavior and provide a framework for precisional targeted therapy.
We review the relevant genetic and epigenetic data in meningiomas and highlight associations with clinical features.
HISTOPATHOLOGICAL CLASSIFICATION OF MENINGIOMAS
The revised 2016 WHO classification of the CNS categorizes meningiomas into 15 different variants [19]. The heterogeneity of their histopathological features is the most interesting characteristic of meningiomas. The 15 variants are classified into three histologic grades (Fig. 1). The WHO grade I (benign) includes nine variants, and the most frequent are meningothelial, fibrous, and transitional variants. Psammomatous, angiomatous, microcystic, secretory, lymphoplasmacyte-rich, and metaplastic variants are also included in grade I [19]. The WHO grade II and III both include three variants [19]. Atypical, chordoid, and clear cell variants are included in grade II, whereas anaplastic, papillary, and rhabdoid variants are included in grade III [19]. Each variant is diagnosed according to their characteristic histologic morphology. However, many meningiomas show mixed histology in the same tumor, so are diagnosed by the dominant histology. Atypical and anaplastic meningiomas have a set of morphological criteria. Atypical meningiomas are diagnosed either by the presence of 4–19 mitoses per 10 high-power fields (HPF) or by evidence of brain invasion, or by showing at least three of five of the following features: high cellularity, small cells with high nuclear-to-cytoplasmic ratio, sheeting (uninterrupted patternless or sheet-like growth), prominent nucleoli, and spontaneous necrosis [19]. Anaplastic meningiomas are diagnosed either by markedly elevated mitotic activity (≥20 mitoses per 10 HPF) or by overtly malignant morphology such as carcinomatous, sarcomatous, and melanomatous cytology [19].
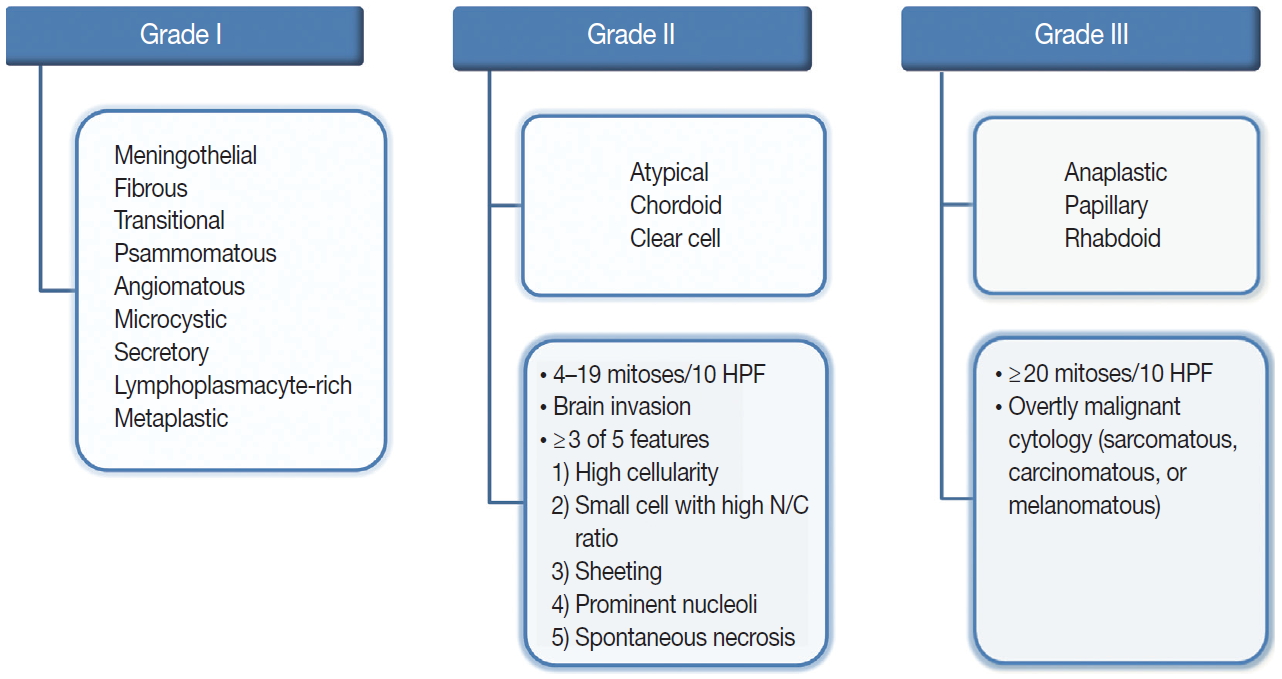
Overview of the 2016 World Health Organization classification system for grading meningiomas. HPF, high-power fields; N/C, nuclear/cytoplasmic.
Although the current histologic grade is valuable in the prognostication of meningiomas, there is still considerable unreliability in predicting clinical behavior and the risk of recurrence and, moreover, determining clinical management. Recently, the revised 2016 WHO classification of the CNS consents to integrated diagnosis, incorporating molecular findings in the diagnosis of some brain tumors, such as gliomas, but not yet meningiomas [20]. The integration of essential molecular profiles in current diagnosis may refine the pathologic classification of meningiomas.
GENETIC ALTERATIONS IN MENINGIOMAS
In the 1970s, the loss of chromosome 22 was first established by a fluorescence technique as a recurrent genetic alteration in meningiomas [21]. Several rare familial syndromes predisposing individuals to meningioma were identified, including neurofibromatosis type 2 (NF2) [22]. In the 1990s, the mutation of the neurofibromin 2 (NF2) gene on chromosome 22 was identified as a major driver, with an incidence of 40%–60% in sporadic meningiomas [23]. Since then, efforts to discover other candidate genes of NF2 wild-type meningiomas have continued, but no critical genetic driver was found until 2013. Powered by the whole-genome sequencing technique, a number of recurrent genetic drivers have been uncovered, such as tumor necrosis factor receptor-associated factor 7 (TRAF7), Kruppel-like factor 4 (KLF4), v-Akt murine thymoma viral oncogene homolog 1 (AKT1), and smoothened frizzled class receptor (SMO) [24,25]. In 2016, the mutation of the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene was also identified as a repetitive driver in some meningiomas (Fig. 2) [26].
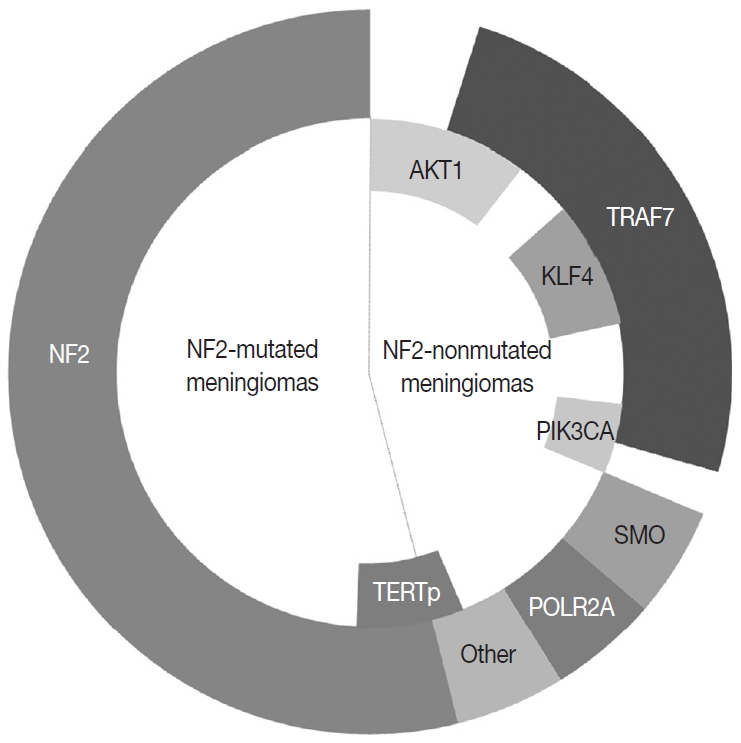
The most common genetic alterations found in meningiomas.
We first review the genetic status of meningiomas in terms of cytogenetic alterations, followed by germline mutations along with familial syndrome, and then somatic mutations.
CYTOGENETIC PROGRESSION OF MENINGIOMAS
Cytogenetic studies provide insight into the genetic status of tumors and launch studies for candidate driver genes because a deletion or duplication in a large fraction of chromosome affects a number of genes within these regions. In meningiomas, the loss of chromosome 22 was found early in the cytogenetic era [21] and brought about the identification of NF2 (located on chromosome 22q) as a powerful driver gene [27].
The loss of chromosome 22q is the most common cytogenetic alteration in meningiomas, found in 60%–70% of all meningiomas [28]. The frequency of this alteration increases with tumor grade, with an incidence of 50% and 75%–85%, in benign and atypical or anaplastic meningiomas, respectively [29]. Except for this loss of 22q, benign meningiomas are typically stable at the cytogenetic level [29]. Unusually, angiomatous meningioma harbors multiple polysomies including chromosome 5, 13, and 20, but do not manifest aggressive behavior [30].
In general, the cytogenetic alterations accumulate with increasing tumor grades, therefore atypical and anaplastic meningiomas show complex cytogenetic profiles (Fig. 3) [29,31]. The losses of chromosome 1p, 14q, and 9p are major additional alterations found in higher-grade meningiomas, and the losses of chromosome 6q, 10, and 18q or the gain of 1q, 9q, 12q, 15q, 17q, and 20q are also noted [28,29,32]. The loss of 1p is the second most frequent alteration and related with a higher recurrence rate [33]. The candidate genes on this chromosomal arm include TP73, CDKN2C, RAD54, EPB41, GADD45A, and ALPL [34-37]. Similarly, the loss of 14q is common in higher-grade tumors, resulting in the inactivation of genes located there, such as NDRG family member 2 (NDRG2) and maternally expressed gene 3 (MEG3), and are associated with poor prognosis [38,39]. Codeletion of 1p and 14q is also reported with early recurrence and progression of tumors, suggesting an independent prognostic factor for the WHO grade [40].
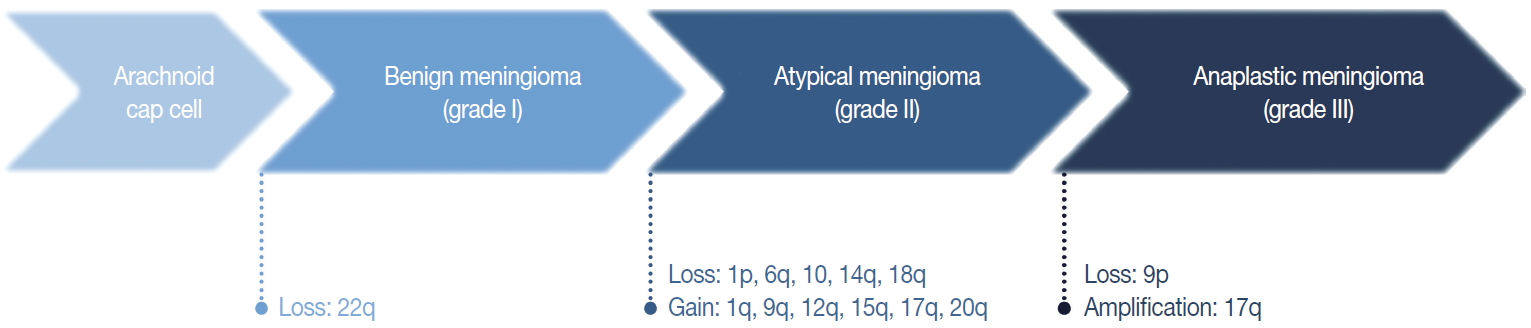
Cytogenetic changes during initiation and progression of meningiomas.
The loss of chromosome 9p is strongly associated with malignant progression to anaplastic meningioma, predicting short survival and a worse prognosis [29,33]. This chromosomal arm harbors several important genes regulating cell cycle and apoptosis, such as cyclin-dependent kinase inhibitor 2A (CDKN2A) and cyclin-dependent kinase inhibitor 2B (CDKN2B) [41]. The amplification of 17q is also found in anaplastic meningioma [42]. Interestingly, recurrent and progressive meningiomas exhibit a more frequent loss of 1p, 14q, and 22q than de novo higher-grade tumors.
GERMLINE ALTERATIONS AND FAMILIAL SYNDROMES PREDISPOSING MENINGIOMAS
Although the majority of meningiomas arise sporadically, several rare familial syndromes (Table 1) increase the risk of meningioma development [22,43]. Understanding these syndromes will help the management of patients with familial history, as well as give the basis for further clarification of meningioma tumorigenesis.
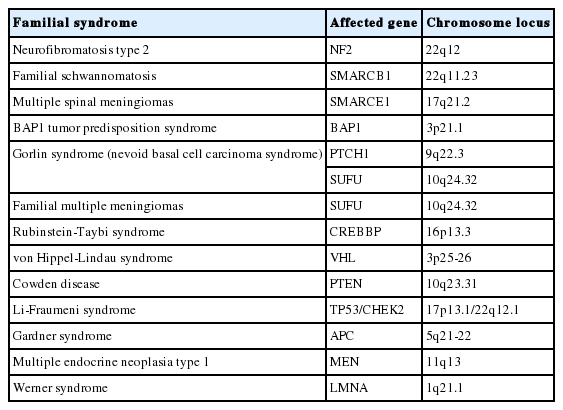
Familial syndromes associated with meningiomas
Neurofibromatosis type 2
NF2 is the most well-known familial syndrome, which is caused by the germline mutation of the NF2 gene on 22q12.2 and characterized by the development of multiple benign tumors in the nervous system, such as vestibular schwannoma and meningioma [44]. Over 50% of individuals in this syndrome manifest at least one meningioma in their lifetime, with a mean age of 30 years [22,45]. The type of mutation corresponds to the risk and severity of meningioma, a truncating mutation by frameshift or nonsense is correlated with a larger tumor burden and an early onset of meningioma rather than a nontruncating mutation by missense or splice-site [45]. Most of the meningiomas in an NF2 disorder background present a fibrous or transitional phenotype and are generally more aggressive than sporadic tumors [43,46].
Familial syndromes associated with the SWI/SNF complex: SMARCB1 and SMARCE1
The switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex regulates gene expression by nucleosome restructuring and is composed of 10–15 subunits: the ATPase subunits (SMARCA2 or SMARCA4), the conserved core subunits (SMARCB1, SMARCC1, and SMARCC2) and additional complex-specific variant subunits (e.g., SMARCE1) [43]. Genetic aberrations in these subunits are associated with a variety of tumors, and the germline mutations of SMARCB1 and SMARCE1 are found in familial syndrome with risk of meningiomas [47,48]. Germline mutation of the SMARCB1 gene on 22q11.23 causes several hereditary conditions, such as rhabdoid tumor predisposition syndrome (e.g., atypical teratoid/rhabdoid tumor [AT/RT] in CNS) [49], schwannomatosis [50], and Coffin-Siris syndrome [51]. Among these conditions, about 5% of individuals with schwannomatosis develop meningiomas [52], and the remaining two conditions have no significant relation to meningiomas. This is because the type and location of the mutations within the same gene are significantly different from each other, which has an influence on the phenotype of each condition [53]. Schwannomatosis harbors a nontruncating mutation at the beginning or end of the SMARCB1 gene that presents as a benign tumor predisposition syndrome, whereas AT/RT is a highly aggressive malignant tumor in pediatrics and associated with a truncating mutation in the central exons or whole gene [52,53]. Although Coffin-Siris syndrome associated with a nontruncating mutation in exon 8 or 9 is a developmental disorder without the risk of tumors, a point mutation (p.Arg377His) in the SMARCB1 gene was reported as a recurrent somatic mutation in one study [51,54]. SMARCB1 is closely located (6 Mb apart) to NF2 on chromosome 22, and co-mutation of these two genes has been described during the tumorigenesis of meningiomas [47]. Germline mutation of SMARCE1 gene on 17q21.2 was identified in families with multiple spinal meningiomas, and later this mutation was also found in individuals with intracranial meningiomas [48,55]. Almost all SMARCE1 mutations are truncating with loss of function mutations and present a specific clear cell morphology [55].
BAP1 tumor predisposition syndrome
Germline mutation of the BRCA1-associated protein 1 (BAP1) gene on 3p21.1 is a genetic cause of BAP1 tumor predisposition syndrome (BAP1-TPDS), which is susceptible to various neoplasms, including uveal and cutaneous melanomas, pleural and peritoneal mesotheliomas, renal cell carcinoma, and mesothelioma [56]. Affected individuals initially present with multiple skin-colored or light red dome-shaped papules, so-called BAP1-inactivated nevi/melanocytoma of the skin and develop meningiomas by the time they reach 50 years [57]. Meningiomas in BAP1-TPDS tend to have high-grade rhabdoid morphology and aggressive clinical behavior [56]. BAP1 encodes a ubiquitin carboxyl-terminal hydrolase 1, which is involved in the regulation of transcription factor chromatin modification as a part of the polycomb repressive complex (PRC), and response to DNA damage by interacting with an important tumor suppressor, BRCA1 [56]. Currently, a BAP1 antibody is available for immunohistochemistry (IHC) and the loss of BAP1 expression on IHC is well correlated with the BAP1 mutation [56].
Gorlin syndrome (Nevoid basal cell carcinoma syndrome): PTCH1 and SUFU
Several genes in the sonic hedgehog (SHH) signaling pathway are relevant to nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, affecting multiple organ systems by nontumorous or tumorous conditions [44]. Together with diverse craniofacial and skeletal abnormalities, multiple basal cell carcinomas and medulloblastomas are presented [44]. The SHH pathway plays a critical role in embryonic development, and then it is strictly regulated in adult tissue [58]. Aberrant SHH signaling is reported in various solid cancers [58]. Germline mutations of the human homolog of the Drosophila patched gene (PTCH1) on 9p22.32 and suppressor of fusion (SUFU) gene on 10q24.32 increase the risk of meningiomas in this syndrome [59,60]. Normally, the PTCH1 protein maintains this pathway in an inactivated state by retaining SMO. The inactivating germline mutation of PTCH1 in NBCCS leads to aberrant activation of the SHH pathway, which is responsible for meningioma development [61]. Another germline mutation of the downstream factor, SUFU, is also infrequently found in NBCCS with pediatric medulloblastoma. A missense mutation of SUFU (p.Arg123Cys) can be found, although rarely, in families with hereditary multiple meningiomas [61].
SOMATIC MUTATIONS IN MENINGIOMAS
NF2-mutated meningiomas
Genetic alterations of the NF2 gene on 22q12.2 are found in up to 60% of sporadic meningiomas and identified not only in benign but also in higher-grade tumors, suggesting an initial role of tumorigenesis in meningiomas [28,62]. Around 60% of meningiomas with monosomy 22 also harbor the second allelic mutation in the remaining NF2 gene, compatible with the twohit hypothesis of the tumor suppressor gene [28,63]. Truncated or nontruncated proteins are made from the NF2 mutation [64]. The protein Merlin, encoded by the NF2 gene, is a member of the protein 4.1 family and keeps the original cell shape by linking membrane proteins to the cytoskeleton [64]. Therefore, NF2 mutant meningiomas tend to exhibit more of a mesenchymal phenotype, such as fibrous or transitional rather than meningothelial histology [28]. The cells with impaired intercellular adhesion due to defected merlin, lose the inhibitory function on contact-mediated cellular proliferation [64]. Merlin is also involved in several signaling pathways, including Hippo, Notch, and mammalian target of the rapamycin (mTOR), so the loss of this protein results in the dysregulation of cell proliferation, growth, and motility [28,65]. The finding that NF2-mutated meningiomas harbor more genetic alterations than the NF2-nonmutated tumor, even within the same benign grade, gives the impression that chromosomal instability is increased with the presence of the NF2 mutation [66].
NF2-nonmutated meningiomas
About 40% of sporadic meningiomas occur in context with no NF2 mutation, that is, these tumors are driven by other genetic alterations [6]. In the last decade, next-generation sequencing has elucidated a number of recurrent genetic alterations in NF2-nonmutated meningiomas, typically including TRAF7, KLF4, AKT1, SMO, PIK3CA, and RNA polymerase II subunit A (POLR2A) [22,24,25]. These somatic mutations are usually found in grade I meningiomas and mostly do not coexist with NF2 mutations or monosomy 22 [22]. TRAF7 mutations present alone or co-occur with KLF4, AKT1, or PIK3CA mutations, whereas SMO and POLR2A mutations are usually mutually exclusive (Fig. 2) [22,67]. In addition, the somatic mutation of the telomerase reverse transcriptase (TERT) promoter is revealed as a notable factor in the prognosis of meningiomas [22]. Here we detail the relevant somatic mutations one by one (Table 2).
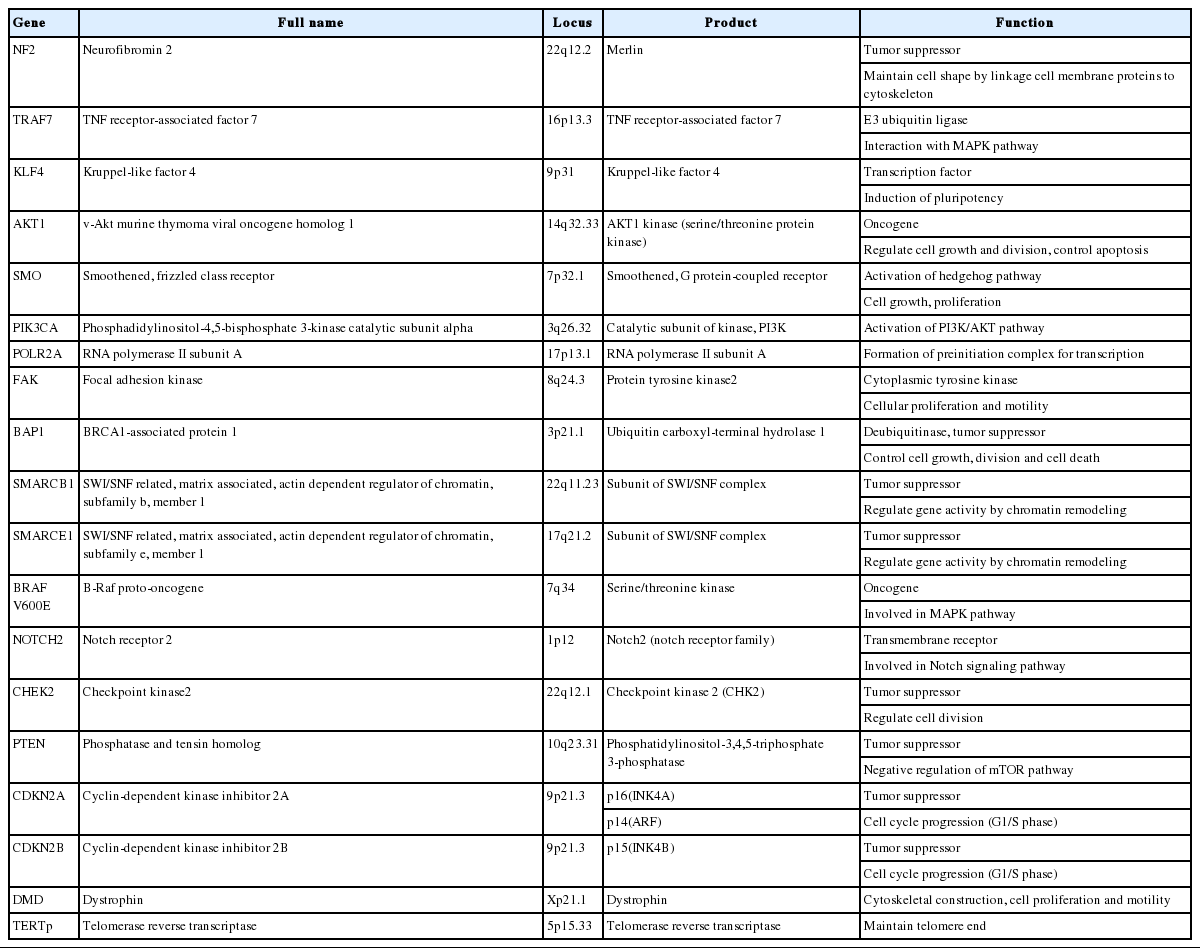
Genes associated with meningiomas
TRAF7 mutations
The somatic mutation of the TRAF7 gene on 16p13.3 is the most frequent genetic aberration observed in meningiomas without an NF2 mutation and found in 15%–25% of sporadic meningiomas [24]. After the TRAF family was first identified as a signal adaptor of the tumor necrosis factor (TNF)-receptor superfamily, diverse receptor families engaging TRAFs for adaptor molecules have been found [68]. In addition, most TRAFs play a role as E3 ubiquitin ligase, activate downstream signaling pathways, and lead to the activation of mitogen-activated protein kinase (MAPK), nuclear factor-κB, and interferon-regulatory factor [68,69]. TRAF proteins transport various stimuli into the cell and engage in multiple physiologic processes, such as embryonal development, regulation of immunity, and stress response with tissue homeostasis [68]. The role of TRAF proteins in the pathogenesis of multiple diseases has been established, and aberrant TRAF7, the last member of the family, was identified as an important genetic driver of meningiomas [6]. TRAF7 mutations are mainly confined to exons 13–20, encoding seven WD40 repeats and approximating the domain through which TRAF7 interacts with other factors [24]. TRAF7 mutated meningiomas usually show meningothelial and benign histology, but also present with higher-grade tumors [24]. TRAF7 mutations frequently co-occur with other somatic mutations, including KLF4, AKT1, and PIK3CA mutations, but are mutually exclusive to NF2 or SMO mutations. Depending on what it is a coexisting mutation, it will affect the prognosis of TRAF7 mutant meningioma [24,70]. Interestingly, almost all secretory meningiomas carry mutations in both TRAF7 and KLF4 genes [70].
KLF4 mutations
The KLF4 gene on 9q31 encodes a transcription factor belonging to a large Kruppel-like factor family, which is involved in many physiologic processes including proliferation, apoptosis, development, and pluripotency [71]. It promotes the reprogramming of differentiated adult cells to pluripotent stem cells, which might give a chance to driving tumors [71]. The only recurrent mutation of KLF4 is a missense mutation at codon 409, which is present in the DNA-binding domain [72]. The substitution of lysine to glutamate (p.Lys409Gln) in that site affects the interaction with DNA target sequence and gene expression [72]. About one-fourth of NF2-nonmutated meningiomas harbor a KLF4 mutation, corresponding to 9%–12% of all meningiomas [24]. KLF4 mutations occur frequently with TRAF7 mutation and exclusively with NF2 and AKT1 mutations [24,70]. Nearly all secretory meningiomas have KLF4 and TRAF7 co-mutations [70]. Secretory meningiomas have cytokeratin-positive globules, which is related with the regulatory role of KLF4 to cytokeratin 4 and 19 [72].
AKT1 mutations
AKT1 on 14q32.33 encodes the AKT1 kinase (serine/threonine-protein kinase), which regulates cell proliferation, growth, and survival by phosphorylation of downstream factors in the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signaling pathway [25,73]. The somatic mutation at codon 17 (p.Glu17Lys) causes an overactive AKT1 protein and constitutive activation of this pathway and is then related to uncontrolled cell proliferation [73]. The AKT1 mutation is found in 7%–12% of grade I meningiomas and is rare in grade II or III [24]. More than half of the AKT1 mutated tumors also harbor mutations in TRAF7 but not in NF2, KLF4, and SMO [24,25]. The AKT1-mutant meningiomas present predominantly meningothelial histology, anterior skull base location, and reduced time to recurrence [74]. Moreover, a somatic mutation in AKT1 (p.Glu17Lys) is also found in Proteus syndrome, a rare disorder characterized by overgrowth of the bones, skin, and other tissues, and some of these develop meningiomas [75].
SMO mutations
As with the AKT1 mutation, the SMO mutation results in uncontrolled activation of the SHH signaling pathway, which then leads to the initiation and growth of meningioma [24,25,60]. The SMO gene on 7q32.1 has two mutation hotspots, more frequently p.Leu412Phe and p.Trp535Leu mutations occur [76]. SMO mutations are found in 1%–5% of NF2-nonmutated meningiomas and are mutually exclusive to TRAF7 [24,25]. Meningiomas with SMO mutation show meningothelial and grade I histology, and are mainly located in the medial anterior skull base near the midline [24,25,77]. A p.Leu412Phe mutation especially reported in olfactory groove meningioma is associated with larger tumor volume and high risk of recurrence [76].
PIK3CA mutations
PI3K is a member of the PI3K/AKT/mTOR signaling pathway, encoded by the PIK3CA gene [26] on 3q26.32. On mutation, PI3K constitutively phosphorylates downstream factor, AKT, and leads to oncogenic activation of this pathway [26]. PIK3CA mutation is found in 4%–7% of meningiomas, in exclusive fashion with NF2, AKT1, and SMO, but often coexists with the TRAF7 mutation [26]. The PIK3CA mutant meningiomas mostly present meningothelial or transitional histology of WHO grade I and locate at the skull base [26,78]. Out of several identified PIK3CA mutation hotspots, p.His1047Arg and p.Glu545Lys are rarely found in WHO grade II or III, suggesting their association with poor prognosis [78].
POLR2A mutations
The POLR2A gene on 17p13.1 encodes DNA-directed RNA polymerase II subunit A, which is the largest subunit of RNA polymerase II and crucial for the formation of the preinitiation complex [79]. Mutations of the POLR2A gene, mainly p.Gly-403Lys or p.Leu438_His439del at exon 7, can affect gene transcription [79]. The POLR2A mutation is found in 6% of meningiomas, exclusively from other mutations [79]. POLR2A mutated meningiomas present meningothelial histology and arise from the tuberculum sellae of the skull base [79]. They are almost all WHO grade I and have a low risk of recurrence [79].
FAK mutations
The focal adhesion kinase (FAK) gene on 8q24.3 encodes a cytoplasmic protein tyrosine kinase, which regulates several cell mechanisms involved in cell growth, proliferation, survival, and motility [80,81]. FAK overexpression has been found in several advanced solid cancers and is a potential therapeutic target [80]. Poulikakos et al. [80] reported a relationship between merlin and the FAK pathway in cancer; merlin negatively regulates the phosphorylation of FAK and inhibits the activity of FAK [81]. Upregulation of FAK is found in some meningiomas and related to a higher grade [82].
BAP1 mutations
As mentioned previously, BAP1 mutations were first reported in a distinct subset of rhabdoid meningiomas as a germline mutation in BAP1-TPDS [57,83]. Some BAP1 mutant meningiomas have no germline mutation, indicating that the somatic mutation of BAP1 also occurs [56]. But the difference in the clinical features between germline and somatic mutations is still unknown, in contrast to other BAP1-associated tumors [56]. Germline mutation is more commonly found in BAP1-mutant meningioma [56]. BAP1 mutation is also related to early tumor recurrence [56]. A wide span of BAP1 gene alterations are detected by BAP1 IHC, which may be a useful surrogate marker for the loss of the BAP1 protein, and is important to the risk stratification of meningiomas showing a rhabdoid morphology [56,57,84].
SMARCB1 and SMARCE1 mutations
As with the BAP1 gene, the somatic mutations of SMARCB1 and SMARCE1 are also found in a few sporadic meningiomas without an inherited background [54]. Generally, these mutations of subunits in the SWI/SNF complex are more frequently found in higher-grade meningiomas and so present poor prognosis [85]. Again, the loss of SMARCE1 expression in IHC is a specific marker for the diagnosis of clear cell meningiomas. Spinal or cranial clear cell meningiomas present moderately aggressive features with a high risk of recurrence [55,86].
BRAF V600E mutations
Somatic mutations of the B-Raf proto-oncogene (BRAF) gene are found in several tumors, including malignant melanoma and papillary thyroid carcinoma [87]. BRAF is located on 7q34 and encodes a serine/threonine kinase of the MAPK pathway, which tightly regulates cell growth and division [87]. The point mutation at codon 600 produces constitutively activated BRAF kinase, that leads to the activation of the downstream pathway and uncontrolled cell proliferation [87]. Two cases of BRAF V600E mutant rhabdoid meningiomas have been reported and one of them, presenting an aggressive clinical course, was improved with BRAF inhibitor (dabrafenib) [88-90]. No other histologic variants of the BRAF V600E mutation were found [88].
NOTCH2 mutations
The Notch receptor 2 (NOTCH2) gene on 1p12 encodes a transmembrane protein of the Notch receptor family, which is involved in Notch signaling pathway and has a role in the developmental process [91]. In meningiomas, the overexpression of activated Notch 2 is thought to be associated with tetraploidy and chromosomal instability, but an accurate mechanism has not yet been found [91].
CHEK2 mutations
The mutation of the checkpoint kinase 2 (CHEK2) gene on 22q12.1 is associated with chromosomal instability in meningiomas [92]. CHEK2 encodes checkpoint kinase 2, which is activated when DNA is damaged. Consequently, it is involved in cell cycle arrest, DNA damage repair, or apoptosis, interacting with other proteins, such as tumor protein 53 (p53). Therefore, the loss of function in this protein leads to the development of tumors [92]. The codeletion of the NF2 gene on the same chromosomal band 22q12 is frequently reported and it is associated with a more aggressive behavior [92].
PTEN mutations
Somatic mutation of the phosphatase and tensin homolog (PTEN) gene on 10q23.31 is one of the most common genetic alterations in numerous tumors, including prostate cancer, endometrial cancer, and gliomas [93,94]. PTEN encodes a phosphatidylinositol-3,4,5-triphosphate 3-phosphatase, which negatively regulates the PI3K/AKT/mTOR pathway [94]. A loss of function mutation in PTEN is responsible for the progression to highergrade meningiomas [95].
CDKN2A and CDKN2B mutations
The CDKN2A gene on 9p21.3 encodes the tumor suppressor proteins, p16(INK4A) and p14(ARF) [35]. The p16(INK4A) protein binds to cyclin-dependent kinase 4 (CDK4) and cyclindependent kinase 6 (CDK6), preventing cell cycle progression and cell division. The p14(ARF) protein also controls cell cycle progression in the G1 phase, protecting the p53 protein from degradation [35]. The CDKN2B gene on 9p21.3 encodes the tumor suppressor, p15(INK4B), which also binds CDK4 or CDK6, preventing cell cycle progression [35]. Mutation or deletion of the chromosomal band harboring CDKN2A and CDKN2B is frequently found in a wide range of tumors [35]. In meningioma, these alterations are usually related to anaplastic histology and poor outcome [41].
DMD mutations
Recently, the dystrophin (DMD) gene on Xp21.2–p21.1 has been suggested as a potential prognostic gene and is originally known for its germline mutation in Duchenne muscular dystrophy [96]. DMD encodes dystrophin, a large protein regulating cytoskeletal construction, cell proliferation, and motility [96]. In addition to a typical myogenic function, a role as an oncogenic gene in tumors originating from the neural crest and mesoderm is indicated [96]. An inactivating mutation of DMD is identified in progressive and higher-grade meningiomas and responsible for poor prognosis [96]. The location on the X chromosome partly supports the fact that aggressive meningiomas are common in males, in contrast to the female predominance of benign tumors [96]. DMD mutation is a mutually independent prognostic factor with TERT promoter alteration [96].
TERT promoter mutations
The TERT gene on 5p15.33 encodes a catalytic subunit of the telomerase complex, maintaining telomeres by adding small DNA repeats (TTAGGG) to the end of the chromosome [97]. In most somatic cells, expression of the telomerase complex is repressed, so the telomere is continuously shortened and finally leads to apoptosis [97]. The somatic mutation of the promoter region of the TERT gene, at hotspot C228T or C250T, increases the number of transcription factor binding sites [98]. This upregulates the expression of TERT and results in the constant survival of tumor cells [97]. In several cancers, such as glioma, melanoma, and bladder cancer, the presence of a TERT promoter mutation is usually associated with aggressive behavior and poor survival [99]. Similarly, in meningiomas, the TERT promoter mutation definitely affects the prognosis [100]. The TERT promoter mutation is more frequently identified in higher grades, 1.7%, 5.7%, and 20% in WHO grade I, II, and III, respectively, and related with the time to progression [100,101]. The median time to progression is 10.1 months versus 179 months in individuals with or without the TERT promoter mutation [100]. In addition, this mutation is mainly found in secondary atypical meningioma progressed from benign tumor rather than de novo atypical meningioma, therefore if it is identified in grade I meningioma, it implicates a high recurrence rate and malignant transformation [100,101].
EPIGENETIC ALTERATIONS IN MENINGIOMAS
Abnormal gene expression is crucial for cancer development and is induced primarily by genetic alterations [7]. However, epigenetic alterations that regulate gene expression without changing the DNA sequence are also responsible for the development of cancer [7]. Recently, evidence that epigenetic alterations have a pivotal role in the tumorigenesis and progression of meningiomas has accumulated [7,102]. In addition, it has been suggested that the status of global DNA methylation more accurately reflects tumor behavior and predicts tumor recurrence compared with WHO and Simpson grades [103-105]. Here we summarize the epigenetic alterations of meningiomas, including aberrant DNA methylation, aberration of chromatin remodeling, and abnormal expression of microRNAs (Table 3).
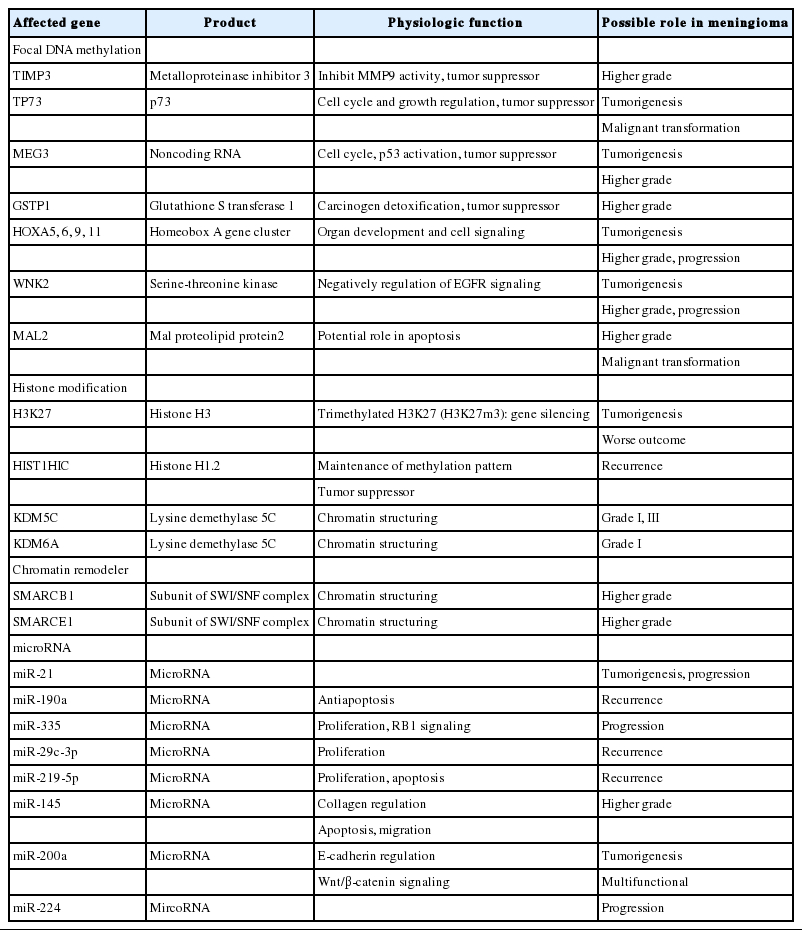
Epigenetic changes in meningiomas
Aberrant DNA methylation
DNA methylation in CpG islands is a well-known epigenetic regulation of gene transcription [106]. Both focal DNA hypermethylation and global DNA methylation profiles have an effect on tumorigenesis [107]. The reliable single genes that are silenced by focal DNA hypermethylation without inactivating mutations in meningiomas include tissue inhibitor of metalloproteinase 3 (TIMP3), tumor protein p73 (TP73), MEG3, glutathione Stransferase Pi 1 (GSTP1), homeobox (HOX) family, WNK lysine deficient kinase 2 (WNK2), and MAL proteolipid protein 2 (MAL2). Hypermethylation of TIMP3 gene on the 22q12.3, inhibits matrix metalloproteinases and results in downregulation of transcription and is associated with higher grades and aggressive behavior [106,108]. Nearly all TIMP3-hypermethylated meningiomas have an allelic loss of 22q [108,109]. Another tumor suppressor gene inactivated by epigenetic hypermethylation is TP73 on 1p36.32, which is found in higher grades, frequently with allelic loss of 1p, and associated with malignant transformation [110]. MEG3 on 14q32.2 encodes an imprinted long noncoding RNA, suppressing tumor cell growth and interacting with the tumor suppressor p53 [111]. MEG3 RNA is considerably expressed in normal arachnoid cells, whereas it is not expressed in meningioma as a consequence of deletion or hypermethylation [39]. The silencing of the MEG3 gene via promoter methylation is more commonly found in higher grades [39]. Hypermethylation of the GSTP1 gene on 11q13.2 is also more frequently found in higher grades [106]. Concordant hypermethylation of HOX genes on 7p15.2, such as HOXA5, HOXA6, HOXA9, and HOXA11, induces silencing and downregulation of target tumor suppressors [112]. The WNK2 gene on 9q22.31 has an inhibitory function on the epidermal growth factor receptor (EGFR) signaling pathway, and the aberrant hypermethylation of this gene results in tumor cell proliferation by relieving inhibition [7,113]. The MAL2 gene on 8q24.12 encodes a transmembrane protein required for the intracellular transport of macromolecules and transcytosis [114]. Hypermethylation of its promoter region and consequent gene silencing are found in higher grade meningiomas [102,114].
Powered by the development of molecular techniques, genomewide methylation profiles are actively investigated in cancer biology. Emerging evidence indicates that global DNA methylation profiles may more precisely predict tumor recurrence and behaviors, and are more useful for the molecular classification of meningiomas [15]. In 2017, the first pivotal study was published by Olar et al. [103] As a result of profiling 140 cases of meningiomas, they recognized two distinct methylation subgroups of meningiomas, naming the prognostically favorable (MM-FAV) and unfavorable (MM-UNFAV) subgroups [103]. They defined a baseline meningioma methylation classifier, which is composed of 238 probes corresponding to hypermethylated CpG loci located on 157 gene lesions [103]. MM-FAV and MM-UNFAV groups show 98 CpG loci for 52 genes and 105 CpG loci for 105 genes, respectively [103]. After further optimization adapting existing prognostic factors, such as WHO and Simpson grades, they derived a 64 CpG loci of meningioma methylation predictor from 238 probes [103]. Each group shows a notably different risk of recurrence [103]. In the same year, Sahm et al. [104] identified two major epigenetic groups, group A and group B, by profiling genome-wide DNA methylation patterns from 497 cases of meningiomas. They further classified the groups into six subgroups, designated into methylation classes (MC), which are associated with distinct patterns of histology, cytogenetic alteration, and mutation [104]. Group A is subdivided into four MC, MC benign 1, 2, 3, and MC intermediate A [104]. Group B is subdivided into two MC, MC intermediate B and MC malignant [104]. They demonstrate that the MCs are well correlated with histologic variant, cytogenetic alteration, and major mutations, and propose that a DNA methylation-based classification system can give a more accurate prognostication of clinical outcomes, such as progression-free survival [104]. Interestingly, they analyzed the potential of this system within the same WHO grade. WHO grade I with MC intermediate had a worse clinical outcome than the average outcome of WHO grade I [104]. Likewise, WHO grade II with MC benign showed a better clinical course than the average of WHO grade II [104]. Accordingly, this system may more accurately define individuals with a higher risk of progression in WHO grade I tumor and individuals with lower risk of progression in WHO grade II, and therefore assist in the precise decision making [104]. More recently, Nassiri et al. [105] announced a DNA methylation-based model for predicting the risk of early 5-year recurrence of meningiomas. They then combined the validated methylome predictor with existing prognostic factors (WHO and Simpson grades) and developed a nomogram calculating a 5-year meningioma recurrence score [105]. It is expected that the meningioma recurrence score will provide information about more personalized clinical outcomes and guidelines for clinical decision making [105].
Aberration of chromatin remodeling
Chromatin remodeling is an important epigenetic regulation of gene expression. It governs the accessibility of the target gene DNA by changing the chromatin structure and exposing or hiding the target region [115]. It is mainly carried out by covalent modification of histone and ATP-dependent regulation of chromatin remodelers [115,116]. Aberration of this chromatin remodeling results in loss of transcriptional regulation of downstream genes and can cause the growth and progression of tumors [115,116]. Deregulation of histone modification and mutations in the members of remodeling complexes are reported in meningiomas [107].
Histone modification
The abnormal modification of histones is reported in many diseases including cancers, although the detailed mechanisms are unclear [117]. Histone proteins not only condense chromatin to form nucleosomes but also participate in the regulation of gene expression through posttranslational modification by methylation, acetylation, phosphorylation, and ubiquitination [118]. It is suggested that modified histones can recruit other proteins, then open or close the chromatin [118]. Among histone members (H2A, H2B, H3, and H4), trimethylation of lysine 27 of histone H3 (H3K27me3) has an important role in the target gene silencing and tumorigenesis of several cancers [115]. Katz et al. [119] reported that meningiomas with the loss of H3K27me3 present more rapid progression and are well correlated with a previously designated DNA-methylation based classification scheme. They suggest that IHC for trimethylation of H3K27 might be useful for the assessment of prognosis particularly in WHO grade II [119]. On the other hand, molecular profiling studies reported mutations of genes associated with histones [120]. An overexpression of the histone cluster H1 family member C (HIST1HIc) gene on 6p22.2 is more frequently found in recurrent meningiomas [120]. It encodes histone H1.2 protein, which interacts with linker DNA and condenses chromatin structure more tightly [120]. It is believed that the H1.2 protein may be engaged in epigenetic regulation of target gene expression by preserving the specific patterns of DNA methylation [120]. In addition, loss of function mutations of KDM5C (on Xp11.22) and KDM6A (on Xp11.3), encoding histone lysine-specific demethylases, affect the function of histones and are also involved in epigenetic regulation in meningiomas [118].
SWI/SNF chromatin remodeler
For the precise expression of genes at the correct time, transcription machinery recruits chromatin remodelers into the chromatin region, where they reconstruct and reposition nucleosome using energy from ATP to obtain a nucleosome-free region for gene expression [115]. The SWI/SNF complex (also known as the BAF complex) is well-known as a chromatin remodeler, and genetic alterations involving various subunits constituting this multimeric complex are found in up to 20% of human neoplasms [121]. It is believed that inactivating mutations of SWI/SNF subunits cause the redistribution of the complex from promoter and enhancer (involved in differentiation) to super-enhancer (involved in selfrenewal, proliferation, and survival) and this imbalance results in tumorigenesis [121]. As previously mentioned, mutations of two core subunits of the SWI/SNF complex, SMARCB1 and SMARCE1, are identified in meningiomas [107,121]. In addition, PRC2, which antagonizes the SWI/SNF complex, is upregulated in high-grade meningioma, therefore loss of function mutations in the SWI/SNF complex plays a critical role in the epigenetic regulation of chromatin in meningioma pathogenesis [122].
MicroRNA expression
MicroRNAs (miRNAs) are important epigenetic mechanisms, which are involved in various physiologic and pathologic processes [123]. MiRNAs are small noncoding RNAs of about 22 nucleotides [107] and regulate the translation of target mRNA by inducing cleavage or degradation of the target mRNA strand and therefore inhibiting translation into proteins [124]. Recently, the role of miRNAs in cancer biology has been actively investigated and considerable genes encoding miRNAs are found in mutational hotspots of tumors or at fragile chromosomal sites [124,125]. Dysregulated miRNA may have either oncogenic or tumor-suppressing functions [124]. Although the miRNA profile of meningiomas has not been so actively studied, an accumulation of evidence implicates the role of miRNAs in the initiation, progression, and recurrence of meningiomas [123]. For example, downregulation of miR-200a and upregulation of β-catenin are found in meningiomas [7,118]. Because miR-200a targets the mRNA of β-catenin, abnormal low expression of miR-200a causes the overexpression of β-catenin and consequently activates the Wnt signaling pathway in meningiomas [118]. Low levels of miR-200a are also associated with the downregulation of E-cadherin via repression of transcription factors, ZEB1 and SIP1 [118]. Accordingly, miR-200a may act as a multifunctional tumor suppressor and the downregulation of miR-200a may promote the development of meningiomas [118,126]. Another study to find candidate miRNA in higher-grade tumors, reported that the downregulation of miRNA-145 is found in higher-grade meningiomas, which is indirectly associated with the overexpression of the COL5A1 gene (encoding collagen type V alpha) and migratory or invasive potential of meningioma cells [118,127]. In addition, a higher expression of miR-21 is found in WHO grade II or III meningioma than in WHO grade I [7,107]. Upregulation of miR-190a and downregulation of miR-29c-3p and miR-219-5p are associated with a significantly high rate of recurrence [7,123]. Overexpression of miR-335 is found in meningiomas and has an effect on tumor cell proliferation, which is associated with the retinoblastoma protein (RB) signaling pathway [7]. MiR-224 is related to the malignant progression of meningiomas and may indicate the prognosis for overall or recurrence free survival [107].
ABERRANT SIGNALING PATHWAYS IN MENINGIOMAS
Most of the genetic alterations impact one or more signaling pathway associated with tumorigenesis and the progression of meningiomas (Fig. 4) [128]. The understanding of aberrant signaling pathways and relevant molecules could provide the basis for the development of corresponding target agents.
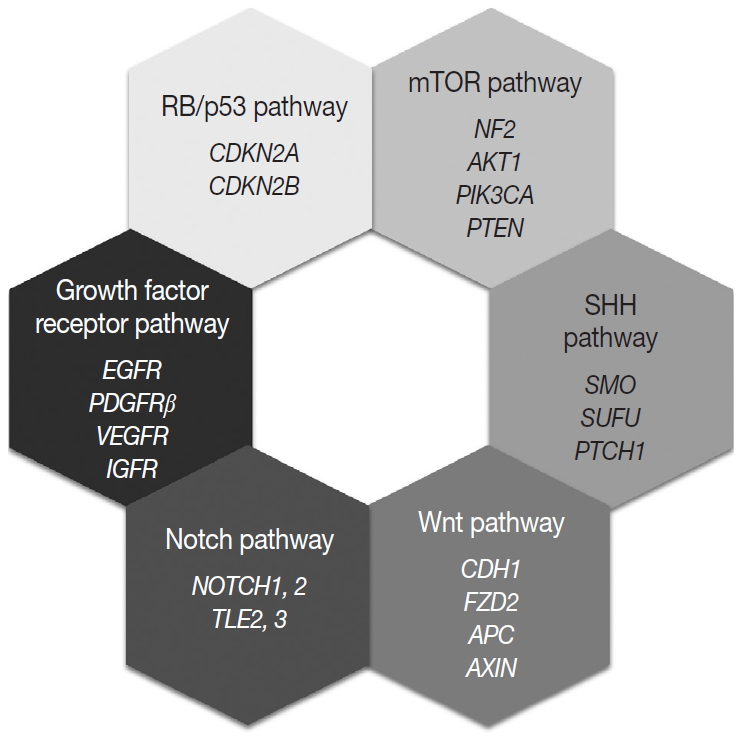
Molecular signaling pathways associated with meningiomas.
mTOR pathway
The aberrant activation of the mTOR pathway has been known to be frequently associated with various human cancers [129]. The key molecule of this pathway, mTOR is a serine/threonine kinase, that constitutes two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [130,131]. Several upstream signaling molecules have an influence on the mTOR signaling pathway, which regulates cell growth, proliferation, survival, and metabolism [131]. Merlin is related to the inhibition of cell proliferation through several pathways, and it is believed that merlin negatively regulates mTORC1 and positively regulates mTORC2 [132]. Consequently, loss of merlin by NF2 mutation causes aberrant overexpression of mTOR and results in the development of meningioma [132]. Another upstream signaling pathway related to mTOR is the PI3K/AKT pathway [129,131]. PI3K phosphorylates and activates AKT, which is an activator of mTORC1 and a substrate for mTORC2 [130]. So activating mutations of PIK3CA and AKT lead to the aberrant activation of the downstream oncogenic mTOR pathway and are found in up to 10% of meningiomas. In addition, PTEN negatively regulates PI3K function, therefore loss of PTEN is also connected to aberrant mTOR activation [130]. Genetic alterations corresponding to the signaling molecules, merlin (NF2), PI3K, AKT, and PTEN, are all identified in a subset of meningiomas [107].
SHH pathway
The SHH signaling pathway is essential for embryonic development [133]. In adults, it is involved and strictly regulated in several key cellular processes, including cell growth, proliferation, stem cell homeostasis, and angiogenesis [107,133]. The SHH ligand binds to the transmembrane receptor, PTCH1, and allows it to relieve its inhibition of SMO [134]. Freely released and activated SMO promotes the translocation of GLI transcription factors in the nucleus and results in the activation of the target gene [128,133]. Genetic alterations of PTCH1 and SMO cause abnormal activation of this pathway and are associated with meningioma tumorigenesis [134]. SUFU is known as a negative regulator of the SHH pathway, and its germline mutation is also reported in meningiomas [107].
Wnt pathway
The loss of E-cadherin expression is found in about 30% of meningiomas and is more frequently found in clinically aggressive and invasive meningiomas [135]. In addition, overexpression and translocation of β-catenin into the nucleus is also found in higher-grade meningiomas [135]. These suggest that Wnt signaling pathway has an important role in the tumorigenesis and progression of meningiomas. One of the receptors of this pathway, frizzled class receptor 2 (FZD2), is highly expressed in meningiomas, which is associated with the activation of the Wnt pathway, overexpression and translocation of cytosolic β-catenin, and the resultant upregulation of oncogenic proteins, such as c-myc and cyclin D1 [107,128,136]. Genetic aberration of genes encoding key molecules of the Wnt pathway, such as adenomatous polyposis coli (APC), E-cadherin (CDH1), and axis inhibition protein (AXIN), are also reported in meningiomas [107,128].
Notch pathway
The Notch signaling pathway is highly conserved and plays an important role in embryonic development by proliferative signaling transduction through four transmembrane receptors, Notch1-4 [137]. When the jagged ligand binds to a Notch receptor, the cleaved intracellular portion of the Notch receptor is translocated into the nucleus and promotes the expression of a transcription factor, hairy and enhancer of split 1 (HES1) [128,135]. HES1 overexpression is found in all grades of meningiomas and is associated with the upregulation of jagged ligand, Notch1, and Notch2 [135]. In contrast, two corepressors modulating HES1 activity, transducin-like enhancer of split 2 (TLE2) and TLE3, are notably upregulated in higher-grade meningiomas [128,135]. As described above, dysregulation of this pathway is associated with tetraploidy and chromosomal instability [137].
Growth factor receptor pathway
Growth factor receptors associated with tumor growth, angiogenesis, and tumor cell migration are implicated for meningioma tumorigenesis, including EGFR, platelet-derived growth factor receptor β (PDGFRβ), vascular endothelial growth factor receptor, and insulin-like growth factor receptor [128,137]. EGFR and PDGFR usually transduce the mitogenic signal via the MAPK signaling pathway, which is reported to be aberrantly activated in meningiomas [135].
RB and p53 pathway
The RB protein regulates the cell cycle progression at the G1/S phase checkpoint by inhibiting the E2F transcription factor [128]. When the mitogenic signal combines cyclin D with either CDK4 or CDK6, it causes phosphorylation of RB and releases the active E2F transcriptional factor, leading to expression of genes associated with the transition from G1 to S phase [128]. The p53 pathway functions as a negative regulator of the RB pathway, so induces the arrest of the cell cycle, repair of damaged DNA, and apoptosis [128,135]. p16(INK4a) and p15 (INK4b) prevent cell cycle progression by inhibiting the CDK4/cyclinD complex, and p14(ARF) connects the two RB and p53 pathways [135]. In fact, genetic alterations of genes encoding these proteins are found in higher-grade meningiomas [128].
CLINICOPATHOLOGICAL FEATURES ASSOCIATED WITH GENETIC ALTERATION IN MENINGIOMAS
The difference in the embryonic origins of the meninges according to site is associated with the frequent histology, location, and recurrent mutations of meningiomas [138]. Meninges of the skull base originate from mesoderm, whereas meninges of the convexity originate from the neural crest [5]. Meningothelial variants are more frequently found in the skull base, whereas fibrous meningiomas mainly develop in the convexity [5,6]. Moreover, genetic alterations of the NF2 gene are preferentially found in the convexity and most other genetic alterations except NF2 are mainly found in skull base tumors (Table 4). The tumor site may also be related to the histologic grade, as the proportion of grade II and III tumors are much higher at the convexity than at the skull base, where grade I meningiomas are more common [6].
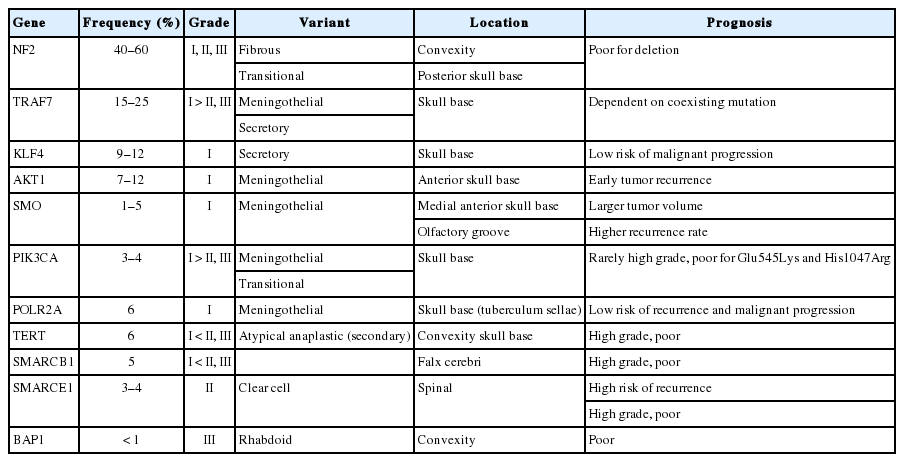
Clinicopathological features related to genetic alteration in meningiomas
AKT1-mutant meningiomas tend to present meningothelial histology and anterior skull base localization, which are associated with early tumor recurrence [74]. SMO-mutant meningiomas also prefer the meningothelial variant at the anterior skull base and olfactory groove region, which tend to have larger tumor volume and higher recurrence rate [76]. Nearly all secretory meningiomas harbor KLF4 mutations with or without TRAF7 mutations but TRAF7 mutations are also recurrent in other histologic variants, mainly the meningothelial variant [70]. POLR2Amutant meningiomas are mainly located at the tuberculum sellae of the skull base and present meningothelial histology [79]. PIK3CA-mutant meningiomas are also located at the skull base and present meningothelial or transitional histology [26,78]. Two types of PIK3CA mutations, p.His1047Arg and p.Glu-545Lys, are more frequently found in higher-grade tumors, which may be associated with poor prognosis [78]. Interestingly, mutation of SMARCE1 is linked to clear cell histology in spinal cord meningiomas [55]. As previously stated, mutations of the TERT promoter obviously has an influence on the prognosis of meningiomas, which is definitely associated with malignant histological progression and time to progression [100]. BAP1 mutations specific for rhabdoid meningioma [56], TP53 mutations, loss of CDKN2A and CDKN2B, and epigenetic inactivation of TIMP3 are all implicated in higher-grade histology and poor clinical outcome [41,106,108]. The deletion of 1p is also associated with the progression of meningiomas [33]. In contrast, genetic alterations of TRAF7, SMO, KLF4, POLR2A, and PIK3CA (except the above two types) are believed to be associated with a relatively low risk of malignant progression and recurrence.
CONCLUSION
The WHO grade and extent of resection are still the most important prognostic factors for meningioma. However, WHO classification does not always predict tumor recurrence or transformation to a higher grade. The molecular characteristics of meningiomas offer profound implications for pathologic classification and allow for the molecular subtyping of meningioma. Furthermore, the discovery of genetic and epigenetic biomarkers of meningioma may assist in the individualized management of precision medicine.
Notes
Author contributions
Conceptualization: Youn Soo Lee, Young Suk Lee.
Data curation: Youn Soo Lee, Young Suk Lee.
Investigation: Youn Soo Lee, Young Suk Lee.
Methodology: Youn Soo Lee.
Supervision: Youn Soo Lee.
Writing—original draft: Youn Soo Lee, Young Suk Lee.
Writing—review & editing: Youn Soo Lee, Young Suk Lee.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding
No funding to declare.