 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 48(2); 2014 > Article
-
Review
Current Concepts in Primary Effusion Lymphoma and Other Effusion-Based Lymphomas - Yoonjung Kim, Chan Jeong Park1, Jin Roh2, Jooryung Huh2
-
Korean Journal of Pathology 2014;48(2):81-90.
DOI: https://doi.org/10.4132/KoreanJPathol.2014.48.2.81
Published online: April 28, 2014
Department of Pathology, Veterans Health Service (VHS) Medical Center, Seoul, Korea.
1Department of Laboratory Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
2Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- Corresponding Author: Jooryung Huh, M.D. Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 138-736, Korea. Tel: +82-2-3010-4553, Fax: +82-2-472-7898, jrhuh@amc.seoul.kr
© 2014 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 18,222 Views
- 182 Download
- 33 Crossref
Abstract
- Primary effusion lymphoma (PEL) is a human herpes virus 8 (HHV8)-positive large B-cell neoplasm that presents as an effusion with no detectable tumor in individuals with human immunodeficiency virus infection or other immune deficiencies. PEL is an aggressive neoplasm with a poor prognosis. PEL cells show diverse morphologies, ranging from immunoblastic or plasmablastic to anaplastic. The immunophenotype of PEL is distinct, but its lineage can be misdiagnosed if not assessed thoroughly. PEL cells usually express CD45, lack B- and T-cell-associated antigens, and characteristically express lymphocyte activation antigens and plasma cell-associated antigens. Diagnosis of PEL often requires the demonstration of a B-cell genotype. HHV8 must be detected in cells to diagnose PEL. In most cases, PEL cells also harbor the Epstein-Barr virus (EBV) genome. Similar conditions associated with HHV8 but not effusion-based are called "extracavitary PELs." PELs should be differentiated from HHV8-negative, EBV-positive, body cavity-based lymphomas in patients with long-standing chronic inflammation; the latter can occur in tuberculous pleuritis, artificial pneumothorax, chronic liver disease and various other conditions. Despite their morphological similarity, these various lymphomas require different therapeutic strategies and have different prognostic implications. Correct diagnosis is essential to manage and predict the outcome of patients with PEL and related disorders.
- PEL is a rare lymphoma that represents approximately 4% of all acquired immunodeficiency syndrome (AIDS)-related lymphomas, as well as 0.1% to 1% of all aggressive lymphomas in HIV-negative immunodeficient patients in regions where HHV8 is not endemic.3 In Korea, PEL comprises approximately 0.1% of non-Hodgkin lymphomas.11,12 Most cases occur in HIV-infected, young to middle-aged, severely immunocompromised, homosexual/bisexual men.1 In the HIV-negative population, PELs have been reported in elderly individuals primarily from regions where HHV8 is endemic, such as Mediterranean regions, but not from Africa, or in solid organ transplant recipients.3,13 Extracavitary PEL accounts for approximately 5% of all AIDS-related lymphomas.5
- HHV8 is a geographically restricted virus. The seroprevalence of HHV8 is >50% in high incidence zones in Africa and parts of the Amazon Basin, 5% to 20% in intermediate incidence zones in Mediterranean countries, and <5% in low incidence zones in North America, Northern Europe, and Asia.14 In Korea, the seroprevalence of HHV8 is 4.93% in the female general population15 and 3.5% in hospitalized children.16 In comparison, the seroprevalence of HHV8 is higher among immunocompromised people; it is 15% among solid organ transplant patients in the United States, and 30% among HIV-infected homosexual/bisexual men.17,18
EPIDEMIOLOGY
- HHV8, like EBV, belongs to the lymphotropic gamma herpesvirus family.13,19,20 HHV8 is transmitted through bodily fluids, such as saliva. Following the primary infection, HHV8 establishes life-long latency in B-cells. During latency, no viral particles are produced, but a set of viral genes that promote cellular proliferation are expressed, which enable the virus to perpetuate in the host B-cell population.13,19 Cytotoxic T-cells normally limit this virally driven cellular proliferation by recognizing viral proteins. Cellular proliferation is unchecked when host immunity is compromised, which increases the chance of malignant clone emergence.5 Viral proteins that have a transforming role in PEL include HHV8/KSHV latent nuclear antigen 1 (LANA-1), v-cyclin, vFLIP (K13), LANA-2 (vIRF-3), vIL-6, K1, kaposin, and K5. These proteins have proliferative, pro-mitotic, anti-apoptotic, pro-inflammatory, and angiogenic functions.5,20 Unknown stimuli can induce HHV8 to transform from its latent state into a lytic state, after which infectious virions are produced and released in saliva. Up to 25% of PEL cells express HHV8 lytic phase genes, which may underlie the reinfection of cells with HHV8, as well as the infection of cells not previously infected.21 During lytic infection, viral genes with pro-inflammatory and angiogenic functions are upregulated, especially those encoding vIL-6, chemokine homologs and their ligands, and transmembrane signaling proteins, which provide further pro-proliferative signals to PEL cells.20
- In PEL, genetic lesions commonly seen in other types of AIDS-related lymphomas are absent, with no MYC or BCL6 gene rearrangements, and no mutations of the RAS oncogene or the TP53 tumor suppressor gene.5,22,23,24 The presence of multiple chromosomal abnormalities suggests that secondary genetic events contribute to neoplastic transformation of PEL cells.24 During latency, HHV8 expresses microRNAs that promote cellular survival, for example, by augmenting nuclear factor-κB activity induced by vFLIP, by targeting IκB, or by preventing cell-cycle arrest through targeting p21 mRNA.5,20
- The vast majority of PEL cells also harbor EBV.3 In HIV-positive patients, PELs are invariably positive for EBV, which are monoclonal in most cases.2 However, the role of EBV in PEL is uncertain because it has a restricted latency pattern with only EBNA1 and EBV-encoded small RNA (EBER) gene expression (latency 1).2,5
ETIOLOGY AND PATHOGENESIS
- In HIV-infected populations, patients with PEL are older and more immunosuppressed than those with Burkitt lymphoma.2 According to Said,2 most HIV-positive PEL patients are in their thirties and have been diagnosed with AIDS, with T-cell counts of <100/mm3. Kaposi's sarcoma lesions are identified in one-third of PEL patients.23 PEL typically presents as a lymphomatous effusion in the pleural, pericardial, or peritoneal cavities, without any extracavitary tumors.1 Typically, a single body cavity is involved. Symptoms include dyspnea and ascites,1,2 and patients usually have B symptoms. While the majority of patients present with no associated mass, a subset of patients develop extension of the pleura or surrounding organs. In approximately one-third of patients,5 the lymphoma disseminates to extracavitary sites, including lymph nodes and bone marrow.
- In extracavitary PEL, tumors occur that have a similar morphology, immunophenotype, and gene expression profile to classical PEL.4,25 Extracavitary PEL commonly involves the gastrointestinal tract, skin, lungs, central nervous system, and lymph nodes.4,22,26 Extracavitary PEL occurs at a slightly earlier stage of HIV infection than PEL, and presents with severe systemic symptoms, generalized lymphadenopathy, and multiple extranodal involvement, with patients frequently showing anemia, hypoalbuminemia, autoimmune thrombocytopenia, and a positive Coomb's test.22
CLINICAL PRESENTATION
- PEL is typically diagnosed on the basis of cytological (cytospin or cell block) preparations of effusion fluid. PEL cells show features that are intermediate between those of immunoblastic and anaplastic large cell lymphomas (ALCLs), with or without plasma cell differentiation (Figs. 1B, 1C, 2C).1,2,22 PEL cells are large, and pleomorphic or monotonous, with marked variation in size (Figs. 1C, 2C). The nuclei of these cells are large, and round to lobated, with prominent nucleoli. These cells contain a variable amount of cytoplasm that is deeply basophilic and often vacuolated. Poorly defined paranuclear hofs are often observed, and anaplastic cells may be detected that resemble Reed-Sternberg cells in classical Hodgkin lymphoma. Similar cells are observed in tissue sections, with tingible body macrophages and numerous mitotic figures with or without a starry-sky appearance (Fig. 2A).1 The marked pleomorphism characteristically seen in cytological specimens may not be apparent in tissue sections.1,27
MORPHOLOGICAL FEATURES
- PEL has a "null" lymphocyte phenotype.27 PEL cells usually express CD45, but lack pan-B-cell antigens (CD19, CD20, CD79a, and surface and cytoplasmic immunoglobulins [Igs]), and T-cell antigens (CD3, CD4, and CD8).1,2,28 These cells are generally positive for markers associated with lymphocyte activation, including CD30, CD38, CD71, epithelial membrane antigen, the human leukocyte antigen DR, and plasma cell differentiation-related antigens (CD138, VS38c, and MUM-1/IRF4) (Figs. 1D, 2E). In a subset of cases, PEL cells are positive for aberrant T-cell markers, such as CD45RO (90%), CD7 (30%), and CD4 (20%), by flow cytometry.29 PEL cells do not usually express Bcl-6,1 are negative for anaplastic lymphoma kinase 1, and usually have a high Ki-67 labeling index. Rare PEL cases with a T-cell phenotype and T-cell receptor gene rearrangement have been reported, and these cases appear to be of T-cell origin.30
- HHV8 must be detected in cells to diagnose PEL. A monoclonal antibody against LANA-1, which is encoded by open reading frame 73, is a highly sensitive and specific marker of HHV8 infection in paraffin-embedded tissue sections (Figs. 1F, 2F).1,2 EBV infection can be demonstrated by in situ hybridization to detect EBER. Lymphoma cells do not express EBV latent membrane protein 1, whereas a subset of cells expresses viral interleukin-6.
IMMUNOPHENOTYPE
- Ig genes are clonally rearranged in PEL, and some cases have rearrangements of both Ig and T-cell receptor genes.2 Somatic hypermutations also occur in PEL. Common genetic abnormalities that are seen in diffuse large B-cell lymphomas are not detected in PEL, including MYC and BCL6 gene rearrangements, and mutations of the RAS oncogene and TP53 tumor suppressor gene.22,23 Gene expression profiling indicates that PEL cells show features of both immunoblasts and plasma cells, but not of germinal center or memory B-cells; MUM-1/IRF-4, CD30, interleukin 10, and vascular endothelial growth factor are upregulated, whereas CD19, CD20, and CD79a/b are downregulated.31 These finding suggests that PEL cells are post-germinal center B-cells or activated B-cells that undergo terminal plasma cell differentiation.22
- Polymerase chain reaction (PCR) detection of HHV8 DNA can demonstrate the presence of the viral genome in tumor tissue (Fig. 1E), which has diagnostic significance.1,2,21 However, the utility of PCR testing for HHV8 DNA in blood and other specimens is limited.21 HHV8 viremia is suggestive of PEL, but cannot be used to definitively diagnose PEL. HHV8 viremia is only observed in 10% to 60% of PEL cases; therefore, PEL cannot be excluded when HHV8 DNA is not detected by PCR.
MOLECULAR GENETIC FEATURES
- When a smear and/or cell block of the effusion shows large malignant cells that are present singly, this can be indicative of undifferentiated carcinoma as well as malignant lymphoma. Cells that have a plasmacytoid morphology and nuclei containing condensed chromatin are likely of a lymphoid origin. However, it may be impossible to rule out carcinoma without performing immunophenotypic studies.
- In patients without a tumor mass but with an effusion that is confirmed to contain cells of a lymphoid origin, PEL must be distinguished from several other types of lymphomas using morphological, immunophenotypic, and clonality analyses, as necessary. PEL and its main differential diagnoses are summarized in Table 1.1,2,9,29,32
- 'Burkitt lymphoma' may present in HIV-positive individuals as a lymphomatous effusion with plasmacytoid differentiation.27 In addition to the distinctive phenotype of Burkitt lymphoma cells (CD20-positive, CD19-positive, BCL6-positive, CD10-positive, BCL2-negative, sIgM-positive, and cIgM-positive), HHV8 negativity and a C-MYC gene rearrangement are also characteristic.2
- 'HHV8-negative effusion-based lymphomas' resembling PELs have been reported. These have been named PEL-like lymphoma, HHV8-unrelated PEL, and body cavity-based high-grade lymphoma.2,8,9,33,34 Patients with HHV8-negative effusion-based lymphomas tend to be elderly (median age 70 years), Japanese (60%), HIV-negative (95.1%), and non-immunocompromised, and there is a slight male predominance (male:female ratio, 3:2). These patients frequently have anti-hepatitis C antibodies (26.5%) and pre-existing medical diseases (>50%) that predispose them to fluid overload, including cirrhosis (hepatitis B, hepatitis C, and alcoholism) and cardiac problems.8,9,33,34 In contrast to PEL, tumor cells in these lymphomas are medium- to large-sized, have an immunoblastic, plasmacytoid, or plasmablastic morphology, and express B-cell-associated antigens (Fig. 3). According to the Hans classification, these tumors have a germinal center B or a mixed germinal center B/activated B-cell signature.9 Moreover, 30% of these tumors are EBV-positive. The prognosis of patients with these types of tumors is more favorable than that of PEL patients.9 Of patients with HHV8-negative effusion-based lymphomas, 70% who were managed with aspiration only and 82.1% who received chemotherapy achieved complete or partial remission,9 with a median survival time of 10 months, which is in sharp contrast to the uniformly poor outcome of PEL patients.
- 'Pyothorax-associated lymphoma (PAL)' presents as a pleural effusion in HIV-negative elderly people, usually males (male: female ratio, 12:3), with a long history of pyothorax or chronic pleuritis resulting from therapeutic artificial pneumothorax or tuberculous pleuritis.35 Most cases have been reported in Japan. PAL develops 20 to 64 years after the onset of tuberculosis and primarily presents as a tumor mass on the body surface. Lymphomatous effusions are rarely detected. PAL cells are morphologically similar to diffuse large B-cell lymphoma, not otherwise specified, with centroblastic or immunoblastic appearances (Fig. 4). All PAL cells express B-cell markers and aberrant T-cell markers, and a fraction express plasma cell markers. PAL cells are HHV8-negative, and, in most cases, EBV-positive.
- 'Plasmablastic lymphoma' is a rare variant of diffuse large B-cell lymphoma. It is characterized by B-immunoblast-like cells with the same immunotype as plasma cells36 and a similar morphology to PEL cells. This condition mainly affects HIV-positive patients, but is also seen in patients with other immunodeficient conditions.36,37 It commonly involves the oral cavity or jaw. Tumor cells can be immunoblastic with prominent central nucleoli, or plasmablastic with abundant basophilic cytoplasm. The immunophenotype of these cells is similar to that of PEL cells, because they are CD20-negative and CD138-positive. Moreover, 50% to 85% and 50% to 70% of plasmablastic lymphoma cases are CD79a-positive and cytoplasmic IgG-positive, respectively.36 EBV infection is found in most plasmablastic lymphoma cases (60-75%), whereas HHV8 is absent.
- 'ALCL' can present as pleural effusion that mimics PEL in rare cases. However, in contrast to PEL, ALCL affects young, HIV-negative individuals. ALCL cells are large lymphoid cells with marked nuclear atypia reminiscent of PEL cells.27,38 ALCL cells have a distinctive phenotype; they are strongly CD30-positive in all cases and frequently positive for epithelial membrane antigen, cytotoxic granule proteins (TIA-1, granzyme, and perforin), CD45, and T-cell-specific markers. In approximately one-third of cases, ALCL cells have a null-cell immunophenotype, despite having a T-cell genotype.38 ALCL cases that have a translocation of the ALK gene still express the ALK protein. ALCL can be distinguished from similar conditions on the basis of the absence of HHV8 and EBV.
- 'Plasma cell myeloma' of a plasmablastic or anaplastic type can mimic PEL. Morphologically, plasmablastic myeloma can be indistinguishable from PEL. Neoplastic plasma cells express CD138 and cytoplasmic IgG, may express CD79a, CD56, and cyclin D1, and may not express CD45 or CD30.39,40,41
- 'ALK-positive large B-cell lymphoma' may exhibit plasmablastic differentiation and mimic PEL. However, these cells do not express CD30,42 are negative for HHV8 and EBV (as determined by labeling for EBER and latent membrane protein 1), and are strongly positive for ALK protein. In most cells, staining for ALK has a granular cytoplasmic pattern that is highly indicative of the t(2;17)(p23;q23) translocation or a CLTC-ALK fusion protein.
- 'Large B-cell lymphoma associated with multicentric Castleman's disease' occurs in patients with or without HIV infection and mainly involves lymph nodes and the spleen.43 It is distinctive from PEL in many ways, although both are associated with HHV8 infection. In large B-cell lymphoma associated with multicentric Castleman's disease, HHV8-positive plasmablasts express high levels of cytoplasmic IgM. In contrast, PEL cells generally do not express cytoplasmic Igs. In large B-cell lymphoma associated with multicentric Castleman's disease, cells harbor non-mutated Ig variable region genes and are derived from CD27- and CD138-negative naive B-cells. By contrast, PEL cells usually express CD138 and harbor hypermutated rearranged Ig genes, suggesting they originate from germinal center or post-germinal center B cells.41,43 Whereas PEL is commonly associated with EBV infection, large B-cell lymphoma associated with multicentric Castleman's disease is not.41,43
- 'Primary pleural effusion post-transplant lymphoproliferative disorder (PTLD)' can occur in transplant recipients.44,45 Most cases of effusion-based PTLD are secondary to widespread solid organ involvement. Primary effusion presentation of PTLD is very uncommon. In this condition, lymphoid cells are monotonous, intermediate to large sized, transformed, have irregular multilobated nuclei and scant cytoplasm, and are located in regions containing abundant karyorrhectic debris. PTLD cells express pan-B-cell markers and are CD30-negative, HHV8-negative, and EBER-positive.
DIFFERENTIAL DIAGNOSIS
- There is no standard treatment for PEL. Attempts have been made to treat PEL with a standard chemotherapy using the CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) regimen; however, the prognosis for patients with PEL remains extremely poor. Their median survival time is <6 months, and most patients die within 1 year of diagnosis.1,2,27 Causes of death include progression of the disease, opportunistic infections, and other HIV-related complications.27
- HIV-associated PEL should be treated with highly active antiretroviral therapy.46 As mentioned above, treatment of PEL patients with the standard CHOP regimen treatment had disappointing result, whereas treatment with bortezomib-containing regimens reportedly improved the survival of PEL patients.47 Chemotherapy should be coupled with immune reconstitution, as well as treatment with interferon-α in combination with azidothymidine.48
- CONCLUSION
- PEL is an HHV8-related lymphoma that typically presents as a malignant serous effusion with no evidence of a solid tumor mass. Extracavitary PEL typically presents as a solid tumor mass in locations such as lymph nodes and the gastrointestinal tract without any effusion. These conditions are often difficult to diagnose owing to the null-cell phenotype of the cells. Other tumors with a similar clinical presentation, morphology, and immunophenotype to PEL should be carefully excluded to properly manage and predict the outcome of PEL patients. HHV8 must be detected in cells to diagnose PEL.
TREATMENT AND PROGNOSIS
- 1. Said J, Cesarman E. Primary effusion lymphoma. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 260-261.
- 2. Said J. Hematopathology of human Immunodeficiency virus (HIV) infection. In: Jaffe ES, Harris NL, Vardiman JW, Campo E, Arber DA, eds. Hematopathology. Philadelphia: Elsevier Saunders, 2011; 867-883.
- 3. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995; 332: 1186-1191. ArticlePubMed
- 4. Chadburn A, Hyjek E, Mathew S, Cesarman E, Said J, Knowles DM. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am J Surg Pathol 2004; 28: 1401-1416. ArticlePubMed
- 5. Cesarman E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 2011; 305: 163-174. ArticlePubMedPMC
- 6. Petitjean B, Jardin F, Joly B, et al. Pyothorax-associated lymphoma: a peculiar clinicopathologic entity derived from B cells at late stage of differentiation and with occasional aberrant dual B- and T-cell phenotype. Am J Surg Pathol 2002; 26: 724-732. PubMed
- 7. Ohsawa M, Tomita Y, Kanno H, et al. Role of Epstein-Barr virus in pleural lymphomagenesis. Mod Pathol 1995; 8: 848-853. PubMed
- 8. Ichinohasama R, Miura I, Kobayashi N, et al. Herpes virus type 8-negative primary effusion lymphoma associated with PAX-5 gene rearrangement and hepatitis C virus: a case report and review of the literature. Am J Surg Pathol 1998; 22: 1528-1537. PubMed
- 9. Alexanian S, Said J, Lones M, Pullarkat ST. KSHV/HHV8-negative effusion-based lymphoma, a distinct entity associated with fluid overload states. Am J Surg Pathol 2013; 37: 241-249. ArticlePubMedPMC
- 10. Ha SY, Choi YL, Kim SJ, Ko YH. Diffuse large B-cell lymphoma associated with chronic inflammation manifested as a soft tissue mass: incidental discovery on histological examination. Korean J Pathol 2011; 45: 417-422. Article
- 11. Yoon SO, Suh C, Lee DH, et al. Distribution of lymphoid neoplasms in the Republic of Korea: analysis of 5,318 cases according to the World Health Organization classification. Am J Hematol 2010; 85: 760-764. ArticlePubMed
- 12. Kim JM, Ko YH, Lee SS, et al. WHO classification of malignant lymphomas in Korea: report of the third nationwide study. Korean J Pathol 2011; 45: 254-260. Article
- 13. Ariza-Heredia EJ, Razonable RR. Human herpes virus 8 in solid organ transplantation. Transplantation 2011; 92: 837-844. ArticlePubMed
- 14. Moore PS, Chang Y. Molecular virology of Kaposi's sarcoma-associated herpesvirus. Philos Trans R Soc Lond B Biol Sci 2001; 356: 499-516. ArticlePubMedPMCPDF
- 15. de Sanjose S, Mbisa G, Perez-Alvarez S, et al. Geographic variation in the prevalence of Kaposi sarcoma-associated herpesvirus and risk factors for transmission. J Infect Dis 2009; 199: 1449-1456. ArticlePubMed
- 16. Han TH, Chung JY, Kim SW. Seroprevalence of human herpesvirus 8 in children in Seoul, Korea. Pediatr Infect Dis J 2005; 24: 476.Article
- 17. Lennette ET, Blackbourn DJ, Levy JA. Antibodies to human herpesvirus type 8 in the general population and in Kaposi's sarcoma patients. Lancet 1996; 348: 858-861. ArticlePubMed
- 18. Jenkins FJ, Hoffman LJ, Liegey-Dougall A. Reactivation of and primary infection with human herpesvirus 8 among solid-organ transplant recipients. J Infect Dis 2002; 185: 1238-1243. ArticlePubMed
- 19. Edelman DC. Human herpesvirus 8: a novel human pathogen. Virol J 2005; 2: 78.ArticlePubMedPMCPDF
- 20. Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 2010; 120: 939-949. ArticlePubMedPMC
- 21. Gantt S, Casper C. Human herpesvirus 8-associated neoplasms: the roles of viral replication and antiviral treatment. Curr Opin Infect Dis 2011; 24: 295-301. PubMedPMC
- 22. Carbone A, Gloghini A. AIDS-related lymphomas: from pathogenesis to pathology. Br J Haematol 2005; 130: 662-670. ArticlePubMed
- 23. Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 1996; 88: 645-656. ArticlePubMedPDF
- 24. Ansari MQ, Dawson DB, Nador R, et al. Primary body cavity-based AIDS-related lymphomas. Am J Clin Pathol 1996; 105: 221-229. ArticlePubMed
- 25. Deloose ST, Smit LA, Pals FT, Kersten MJ, van Noesel CJ, Pals ST. High incidence of Kaposi sarcoma-associated herpesvirus infection in HIV-related solid immunoblastic/plasmablastic diffuse large B-cell lymphoma. Leukemia 2005; 19: 851-855. ArticlePubMedPDF
- 26. Pan ZG, Zhang QY, Lu ZB, et al. Extracavitary KSHV-associated large B-Cell lymphoma: a distinct entity or a subtype of primary effusion lymphoma? Study of 9 cases and review of an additional 43 cases. Am J Surg Pathol 2012; 36: 1129-1140. PubMed
- 27. Patel S, Xiao P. Primary effusion lymphoma. Arch Pathol Lab Med 2013; 137: 1152-1154. ArticlePubMedPDF
- 28. Carbone A, Gloghini A, Larocca LM, et al. Expression profile of MUM1/IRF4, BCL-6, and CD138/syndecan-1 defines novel histogenetic subsets of human immunodeficiency virus-related lymphomas. Blood 2001; 97: 744-751. ArticlePubMedPDF
- 29. Vega F. Primary effusion lymphoma (PEL) and solid variant of PEL. In: Medeiros LJ, Miranda RN, Wang SA, eds. Diagnostic pathology: lymph nodes and spleen with extranodal lymphomas. Altona: Amirsys, 2011; 7: 78-85.
- 30. Coupland SE, Charlotte F, Mansour G, Maloum K, Hummel M, Stein H. HHV-8-associated T-cell lymphoma in a lymph node with concurrent peritoneal effusion in an HIV-positive man. Am J Surg Pathol 2005; 29: 647-652. ArticlePubMed
- 31. Klein U, Gloghini A, Gaidano G, et al. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 2003; 101: 4115-4121. ArticlePubMed
- 32. Ferry JA. Extramodal lymphomas. Philadelphia: Saunders-Elsevier, 2011; 81-132.
- 33. Kang SY, Park CJ, Huh JR, Kang YG, Lee YS, Chi HS. Three cases of primary effusion lymphoma. Korean J Hematol 2003; 38: 136-141.
- 34. Kim YA, Huh J, Yoon SO, et al. Primary effusion lymphoma and human herpesvirus 8-negative effusion-based lymphoma: analysis of 17 cases in Korea. Korean J Pathol 2013; 47(Suppl 1): S100.
- 35. Chan JK, Aozasa K, Gaulard P. DLBCL associated with chronic inflammation. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 245-246.
- 36. Stein H, Harris NL, Campo E. Plasmablastic lymphoma. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 256-257.
- 37. Lee HI, Koo HR, Han EM, et al. Fine needle aspiration cytology of the plasmablastic lymphoma in human immunodeficiency virus (HIV) negative patient: a case report. Korean J Cytopathol 2005; 16: 47-51.
- 38. Delsol G, Falini B, Muller-Hermelink HK, et al. Anaplastic large cell lymphoma (ALCL), ALK-positive. In: Swerdlow SH, Campo E, Harris NL, eds. WHO cclassification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 312-316.
- 39. McKenna RW, Kyle RA, Kuehl WM, Grogan TM, Harris NL, Coupland RW. Plasma cell neoplasms. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 200-213.
- 40. Vega F, Chang CC, Medeiros LJ, et al. Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly identical immunophenotypic profiles. Mod Pathol 2005; 18: 806-815. ArticlePubMedPDF
- 41. Kim Y, Leventaki V, Bhaijee F, Jackson CC, Medeiros LJ, Vega F. Extracavitary/solid variant of primary effusion lymphoma. Ann Diagn Pathol 2012; 16: 441-446. ArticlePubMed
- 42. Delsol G, Campo E, Gascoyne RD. ALK-positive large B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 254-255.
- 43. Isaacson PG, Campo E, Harris NL. Large B-cell lymphoma arising in HHV8-associated multicentric Castleman diseae. In: Swerdlow SH, Campo E, Harris NL, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008; 258-259.
- 44. Ohori NP, Whisnant RE, Nalesnik MA, Swerdlow SH. Primary pleural effusion posttransplant lymphoproliferative disorder: Distinction from secondary involvement and effusion lymphoma. Diagn Cytopathol 2001; 25: 50-53. ArticlePubMed
- 45. Lechapt-Zalcman E, Rieux C, Cordonnier C, Desvaux D. Posttransplantation lymphoproliferative disorder mimicking a nonspecific lymphocytic pleural effusion in a bone marrow transplant recipient. A case report. Acta Cytol 1999; 43: 239-242. PubMed
- 46. Ripamonti D, Marini B, Rambaldi A, Suter F. Treatment of primary effusion lymphoma with highly active antiviral therapy in the setting of HIV infection. AIDS 2008; 22: 1236-1237. ArticlePubMed
- 47. Siddiqi T, Joyce RM. A case of HIV-negative primary effusion lymphoma treated with bortezomib, pegylated liposomal doxorubicin, and rituximab. Clin Lymphoma Myeloma 2008; 8: 300-304. ArticlePubMed
- 48. Ghosh SK, Wood C, Boise LH, et al. Potentiation of TRAIL-induced apoptosis in primary effusion lymphoma through azidothymidine-mediated inhibition of NF-kappa B. Blood 2003; 101: 2321-2327. PubMed
REFERENCES
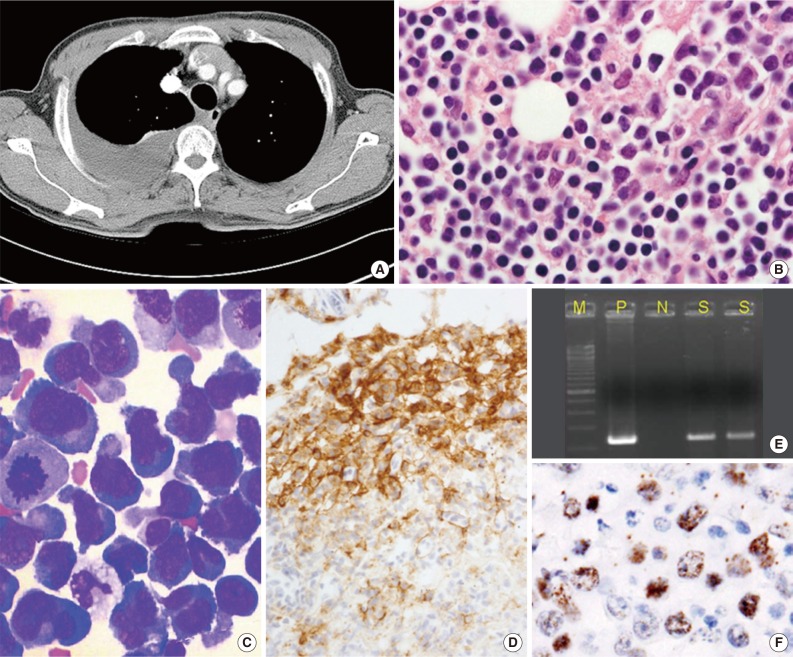
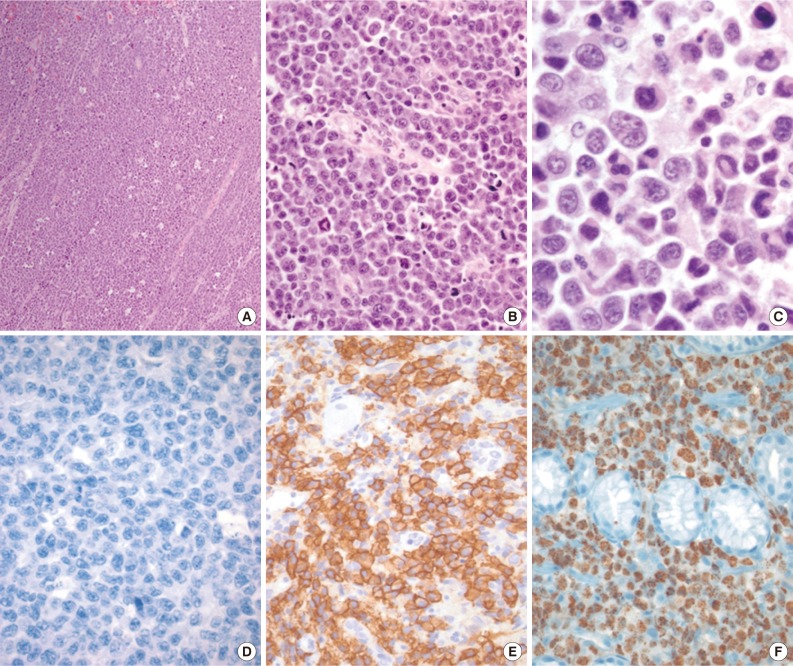
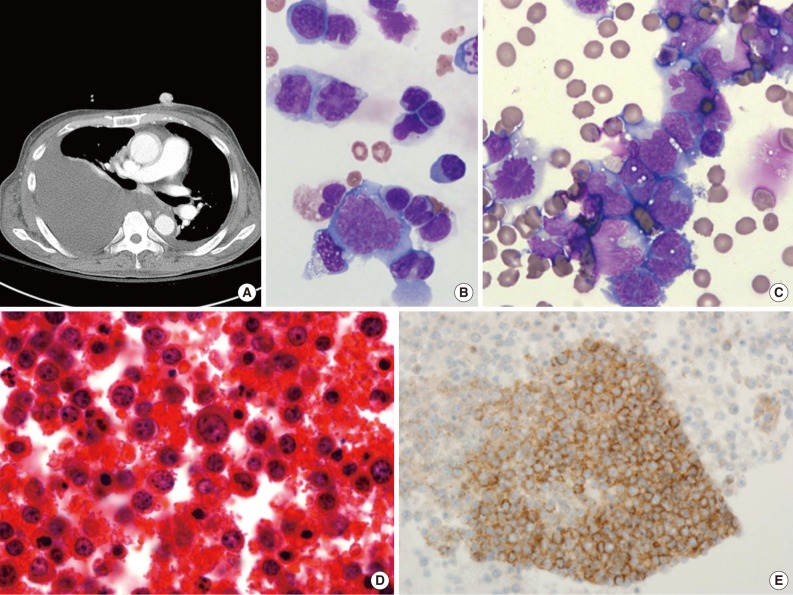
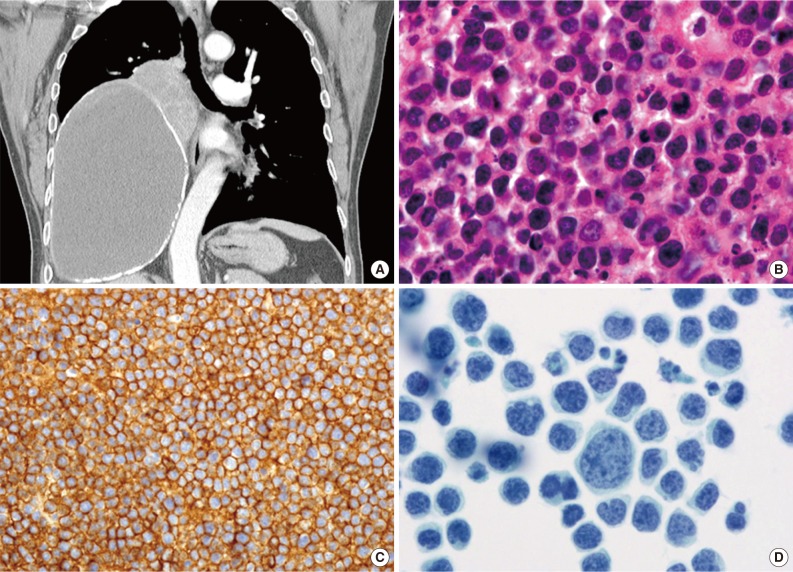
Figure & Data
References
Citations
- Human Herpesvirus-8/Kaposi Sarcoma Herpesvirus in Solid Organ Transplantation: A Narrative Review of Neoplastic Manifestations
Alessandra Mularoni, Andrea Cona, Carlotta Piazza, Francesca Pecoraro, Elda De Vita, Mario Luppi
Current Infectious Disease Reports.2026;[Epub] CrossRef - Real-World Application of the WHO Reporting System for Lymph Node Cytopathology in the Context of the 5th Edition of the WHO Classification of Hematolymphoid Neoplasms
Ana G. Yuil Valdes, Safina Hafeez, Terrance J. Lynn, Joseph D. Khoury
Acta Cytologica.2026; : 1. CrossRef - Primary effusion lymphoma in an HIV-negative patient
I. Gadaev, I. Ilgisonis, D. Budanova, A. Klemeshova, O. Bochkarnikova, O. Antufeeva, I. Sokolova, M. Mingalimov, P. Salamova, E. Fominikh
Vrach.2026; : 91. CrossRef - Imaging Findings of Thoracic Extranodal Lymphoma: A Pictorial Essay
Songa Seo, Soo-yeon Jeong, Mi Sook Lee
Journal of the Korean Society of Radiology.2025; 86(6): 1011. CrossRef - Update: The molecular spectrum of virus-associated high-grade B-cell non-Hodgkin lymphomas
H. Witte, A. Künstner, N. Gebauer
Blood Reviews.2024; 65: 101172. CrossRef - Oncolytic strategy using new bifunctional HDACs/BRD4 inhibitors against virus-associated lymphomas
Jungang Chen, Zhengyu Wang, Tran Phuc, Zhigang Xu, Donglin Yang, Zhengzhu Chen, Zhen Lin, Samantha Kendrick, Lu Dai, Hong-yu Li, Zhiqiang Qin, Michael Lagunoff
PLOS Pathogens.2023; 19(1): e1011089. CrossRef - Primary Effusion Lymphoma: A Timely Review on the Association with HIV, HHV8, and EBV
Chih-Yi Liu, Bo-Jung Chen, Shih-Sung Chuang
Diagnostics.2022; 12(3): 713. CrossRef - Lymphoproliferative disorder involving body fluid: diagnostic approaches and roles of ancillary studies
Jiwon Koh, Sun Ah Shin, Ji Ae Lee, Yoon Kyung Jeon
Journal of Pathology and Translational Medicine.2022; 56(4): 173. CrossRef - Clinical Characteristics and Management of Patients With Concomitant Liver Cirrhosis and Lymphoma: A Systematic Review
Jelena Jelicic, Thomas Stauffer Larsen, Annette Dam Fialla, Zoran Bukumiric, Bosko Andjelic
Clinical Lymphoma Myeloma and Leukemia.2022; 22(11): e981. CrossRef - HHV8-unrelated primary effusion lymphoma: Two case reports and a review of literature
Ryan W. Kendall, Ricky A. Thompson, Christopher P. Garwacki, Alan Z. Skarbnik
Current Problems in Cancer: Case Reports.2021; 4: 100087. CrossRef - Targeting Host Cellular Factors as a Strategy of Therapeutic Intervention for Herpesvirus Infections
Kumari Asha, Neelam Sharma-Walia
Frontiers in Cellular and Infection Microbiology.2021;[Epub] CrossRef - A Rare Case of Extracavitary Primary Effusion Lymphoma in the Bladder and Ureter
Jiankun Tong, Sana Jadallah, William H. Rodgers, Gabriel Jung, Malvina Fulman, Abhisek Swaika
Case Reports in Hematology.2020; 2020: 1. CrossRef - KSHV: Immune Modulation and Immunotherapy
Grant Broussard, Blossom Damania
Frontiers in Immunology.2020;[Epub] CrossRef - Role of Ki 67 Labelling Index as an Adjunct to Histopathological Diagnosis for Grading of CNS Tumours
Priyanka Rai, Chandni Krishnani, Goswami S. S.
Journal of Evolution of Medical and Dental Sciences.2020; 9(16): 1331. CrossRef - Brentuximab vedotin as frontline treatment for HIV-related extracavitary primary effusion lymphoma
Jose D. Sandoval-Sus, Amanda Brahim, Alina Khan, Barbara Raphael, Ali Ansari-Lari, Marco Ruiz
International Journal of Hematology.2019; 109(5): 622. CrossRef - High-dose Therapy and Autologous Hematopoietic Cell Transplantation as Consolidation Treatment for Primary Effusion Lymphoma
Abu-Sayeef Mirza, Bhagirathbhai R. Dholaria, Mohammad Hussaini, Sarah Mushtaq, Pedro Horna, Adharsh Ravindran, Ambuj Kumar, Ernesto Ayala, Mohamed A. Kharfan-Dabaja, Celeste Bello, Julio C. Chavez, Lubomir Sokol
Clinical Lymphoma Myeloma and Leukemia.2019; 19(9): e513. CrossRef - Remission of an HHV8-related extracavitary primary effusion lymphoma in an HIV-positive patient during antiretroviral treatment containing dolutegravir
Laura Campogiani, Carlotta Cerva, Gaetano Maffongelli, Elisabetta Teti, Livio Pupo, Sara Vaccarini, Maria Cantonetti, Alfredo Pennica, Massimo Andreoni, Loredana Sarmati
AIDS Research and Therapy.2019;[Epub] CrossRef - Primary Effusion Lymphoma in a Non-Human Immunodeficiency Virus Patient: A Case Report
Beum Jin Kim, Mi Sook Lee
Journal of the Korean Society of Radiology.2019; 80(4): 810. CrossRef - Pleural effusion in a human immunodeficiency virus‐infected patient
Rafael Martínez‐Girón, Santiago Martínez‐Torre
Cytopathology.2019; 30(6): 673. CrossRef - Case report of a primary effusion lymphoma successfully treated with oral valganciclovir after failing chemotherapy
Juan Marquet, Kyra Velazquez‐Kennedy, Sandra López, Amparo Benito, María‐Jesús Blanchard, Jose Antonio Garcia‐Vela
Hematological Oncology.2018; 36(1): 316. CrossRef - Primary effusion lymphoma in Taiwan shows two distinctive clinicopathological subtypes with rare human immunodeficiency virus association
Bo‐Jung Chen, Ran‐Ching Wang, Chung‐Han Ho, Chang‐Tsu Yuan, Wan‐Ting Huang, Sheau‐Fang Yang, Pin‐Pen Hsieh, Yun‐Chih Yung, Shih‐Yao Lin, Chen‐Fang Hsu, Ying‐Zhen Su, Chun‐Chi Kuo, Shih‐Sung Chuang
Histopathology.2018; 72(6): 930. CrossRef - Effusion‐based lymphoma with morphological regression but with clonal genetic features after aspiration
Meng‐Chen Tsai, Chun‐Chi Kuo, Ying‐Zhen Su, Yen‐Chuan Hsieh, Shih‐Sung Chuang
Diagnostic Cytopathology.2018; 46(8): 685. CrossRef - Biology and management of primary effusion lymphoma
Kazuyuki Shimada, Fumihiko Hayakawa, Hitoshi Kiyoi
Blood.2018; 132(18): 1879. CrossRef - EBV‐associated but HHV8‐unrelated double‐hit effusion‐based lymphoma
Bo‐Jung Chen, David Yen‐Ting Chen, Chun‐Chi Kuo, Shih‐Sung Chuang
Diagnostic Cytopathology.2017; 45(3): 257. CrossRef - HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms
Amy Chadburn, Jonathan Said, Dita Gratzinger, John K. C. Chan, Daphne de Jong, Elaine S. Jaffe, Yasodha Natkunam, John R. Goodlad
American Journal of Clinical Pathology.2017; 147(2): 171. CrossRef - Human immunodeficiency virus (HIV) and Epstein-Barr virus (EBV) related lymphomas, pathology view point
Ebru Linke-Serinsöz, Falko Fend, Leticia Quintanilla-Martinez
Seminars in Diagnostic Pathology.2017; 34(4): 352. CrossRef - Anticancer drug-loaded quantum dots engineered polymeric nanoparticles: Diagnosis/therapy combined approach
D. Belletti, G. Riva, M. Luppi, G. Tosi, F. Forni, M.A. Vandelli, B. Ruozi, F. Pederzoli
European Journal of Pharmaceutical Sciences.2017; 107: 230. CrossRef - CD20-negative diffuse large B cell lymphoma: a comprehensive analysis of 695 cases
Jing Li, Shu Zhao, Jingxuan Wang, Jingyu Chen, Wen Wen, Qingyuan Zhang
Tumor Biology.2016; 37(3): 3619. CrossRef - Co-infections and Pathogenesis of KSHV-Associated Malignancies
Suhani Thakker, Subhash C. Verma
Frontiers in Microbiology.2016;[Epub] CrossRef - Pathology of Extranodal Lymphoma
Emily Heckendorn, Aaron Auerbach
Radiologic Clinics of North America.2016; 54(4): 639. CrossRef - 2015 update on the diagnosis and management of neoplastic pericardial disease
Chiara Lestuzzi, Massimiliano Berretta, Witold Tomkowski
Expert Review of Cardiovascular Therapy.2015; 13(4): 377. CrossRef - CD20-negative diffuse large B-cell lymphomas: biology and emerging therapeutic options
Jorge J Castillo, Julio C Chavez, Francisco J Hernandez-Ilizaliturri, Santiago Montes-Moreno
Expert Review of Hematology.2015; 8(3): 343. CrossRef - Primary Effusion Lymphoma: Cytological Diagnosis of a Rare Entity - Report of Two Cases in HIV-Uninfected Patients from a Single Institution
Marta Nicola, Monica Onorati, Chiara Luisa Bianchi, Giuseppe Pepe, Stefano Bellone, Franca Di Nuovo
Acta Cytologica.2015; 59(5): 425. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
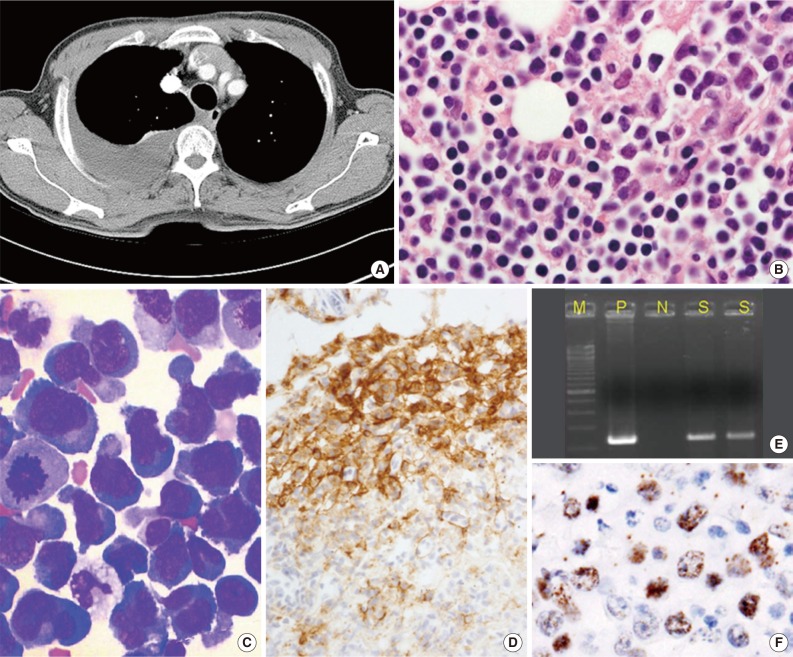
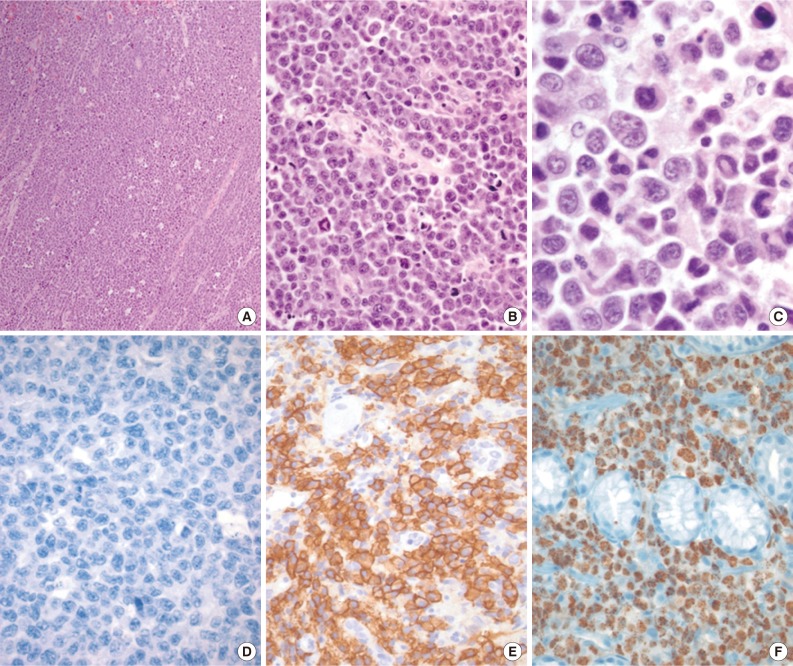
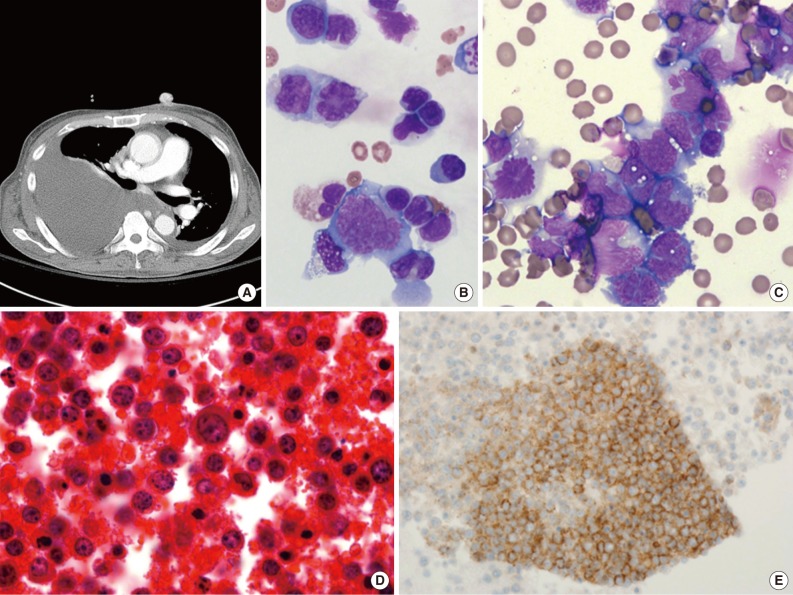
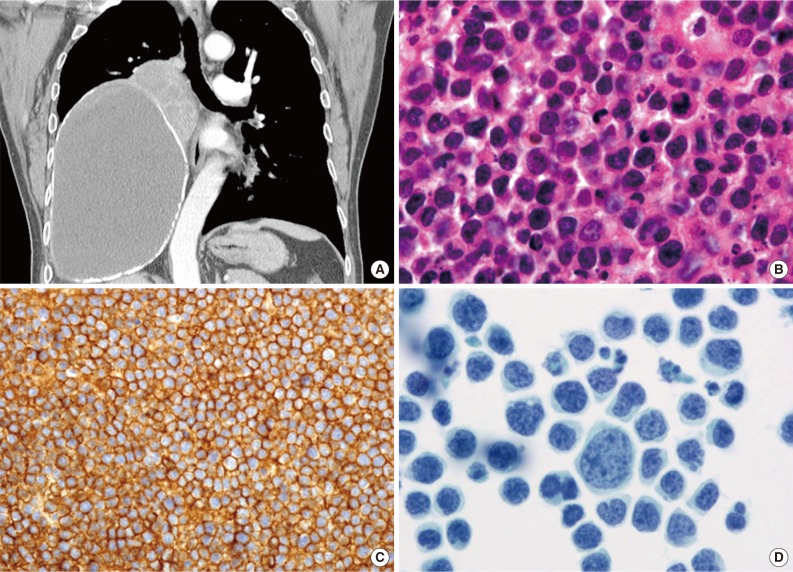




Fig. 1
Fig. 2
Fig. 3
Fig. 4
| Primary effusion lymphoma | Burkitt lymphoma | Plasmablastic lymphoma | HHV8-negative effusion-based lymphoma | Pyothorax-associated lymphoma | |
|---|---|---|---|---|---|
| Demographic characteristics | Young or middle-aged HIV+ (majority) | Endemic: childhood | Median age around 50 yr, mainly adult | Older (median age 70 yr) | HIV-elderly, male predominant, with a long history of pyothorax or chronic pleuritis |
| Sporadic: children, young adults | HIV+ or other immunodeficiency | HIV-(95.1%), and non-immunocompromised anti-HCV (26.5%) | |||
| Immudeficiency-associated | |||||
| Anatomic site | Body cavities | Extranodal; HIV+ patients may present with a lymphomatous effusion with plasmacytoid differentiation | Extranodal; most commonly oral cavity; body cavities - rarely involved | Body cavities | Tumor mass localized in the body surface, and lymphomatous effusion is rare |
| HHV8 | (+) | (-) | (-) | (-) | (-) |
| EBV | (+) (majority) | (+/-) (+) mainly endemic | (+) (60-75%) | (+) (30%) | (+) (most cases) |
| Immuno-phenotype | CD20(–), CD19(–), CD79a(–), CD38(+), CD138(+), CD45(+/–), CD30(+/–) | CD20(+), CD19(+), BCL6(+), CD10(+), BCL2(–), CD30(–), sIgM+, cIgM+ | CD20(–), CD79a+(50-85%), CD38(+), CD138(+), CD19(–), CD45(–), CD30(–/+), cIgG+(50-70%) | CD20(+) (majority), CD19(+), CD138(–/+), CD30(–/+) | CD20(+) (majority), CD19(+), CD138(–/+), aberrant expression of T-cell markers |
| Prognosis | Poor; median survival <6 mo | Endemic, sporadic - highly aggressive but curable | Poor; median survival <1 yr | Variable Median survival 10 mo | 5-yr survival 20-35% |
| IgH | Clonal | Clonal | Clonal | Clonal | Clonal |
| Other genetic features | No recurrent chromosomal abnormalities | C-MYC gene rearrangement | Subset with t(8;14)(q24;q32) or MYC-IgH | Complex karyotypic anomalies in 50% | Complex karyotypic anomalies TP53 mutations (70%) |
HHV8, human herpesvirus 8; HIV, human immunodeficiency virus; HCV, hepatitis C virus; EBV, Epstein-Barr virus; sIg, surface immunoglobulin; cIg, cytoplasmic immunoglobulin; IgH, immunoglobulin heavy chain gene rearrangement.

