 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 46(5); 2012 > Article
-
Case Report
Rosai-Dorfman Disease in the Breast with Increased IgG4 Expressing Plasma Cells: A Case Report - Yoon Jin Cha, Woo Ick Yang, Se Ho Park1, Ja Seung Koo
-
Korean Journal of Pathology 2012;46(5):489-493.
DOI: https://doi.org/10.4132/KoreanJPathol.2012.46.5.489
Published online: October 25, 2012
Department of Pathology, Yonsei University College of Medicine, Seoul, Korea.
1Department of General Surgery, Yonsei University College of Medicine, Seoul, Korea.
- Corresponding Author: Ja Seung Koo, M.D. Department of Pathology, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul 120-752, Korea. Tel: +82-2-2228-1772, Fax: +82-2-362-0860, KJS1976@yuhs.ac
• Received: October 25, 2011 • Revised: December 13, 2011 • Accepted: December 15, 2011
© 2012 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Rosai-Dorfman disease (RDD) can present in any anatomic site, but breast involvement is rarely reported. Recently, a relationship between RDD and IgG4-related sclerosing disease has been suggested. Here we report another case of RDD with overlapping features of IgG4-related sclerosing disease occurring in a right breast of a 62-year-old female. On microscopic examination, the mass demonstrated a characteristic zonal pattern of proliferation of large polygonal histiocytes and lymphoplasma cells with stromal fibrosis. Emperipolesis was observed in histiocytes with abundant cytoplasm, which showed immunoreactivity for S-100 protein and CD68; the diagnosis of RDD was made. Sheets of plasma cells in the fibrotic stroma demonstrated positive reactions for IgG and IgG4. The mean count of IgG4-positive plasma cells was 100.2/high power field, and the ratio of IgG4/IgG was 56.7%. Additional findings of stromal fibrosis and obliteration of preexisting breast lobules suggested overlapping features with IgG4-related sclerosing disease.
- A 62-year-old woman presented with a palpable mass in the right breast. Mammography (Fig. 1A) and ultrasonographic evaluation demonstrated a hyperechoic nodule in the right lower portion. Radiology suggested the diagnostic possibility of a benign lesion such as fat necrosis. A needle biopsy of the breast nodule was performed, and the following diagnosis was made: "prominent aggregation of foamy histiocytes associated with diffuse infiltration of lymphoplasma cells, neutrophils, and lymphoid follicles." The possibility of xanthogranulomatous mastitis, plasma cell mastitis, and the like was suggested. Periodic acid-Schiff staining with prior diastase treatment (D-PAS) and Ziehl-Neelsen staining for identifying microorganisms were all negative. Cytokeratin staining for epithelial lesions was totally negative.
- Six months later, a follow-up sonographic examination showed no changes in the size or echo-density of the nodule. Axillary lymphadenopathy was absent. The skin of the overlying area showed inflammatory change, and the patient underwent surgical excision.
- The excised specimen was a lump of breast tissue. It was firm in consistency, and a 2.8 cm×2.5 cm ill-defined lobulated mass-like lesion was found on sectioning.
- Microscopic examination revealed dense lymphoplasmacytic infiltration in the mass-forming area. Fibrosis and histiocytic infiltrates in a storiform fashion were observed under low-power microscopic examination. Most breast parenchyma was replaced by histiocytes, inflammatory cells, and fibrosis, and these inflammatory cell infiltrates extended into the adjacent fat tissue (Fig. 1B). Diffusely scattered lymphoid follicles were found. Obliterative phlebitis was not present.
- The most prominently recognizable cells were histiocytes with large vesicular nuclei and abundant, clear cytoplasm. Furthermore, some of the histiocytes had intact lymphocytes, plasma cells, and red blood cells within the cytoplasm (Fig. 1C). The infiltrating histiocytes showed strong immunoreactivity for S-100 protein (1:2,000, polyclonal, Dako, Carpinteria, CA, USA) in their nuclei and cytoplasm (Fig. 1D). CD68 (1:100, clone PGM1, Dako) on the histiocytes demonstrated a granular cytoplasmic staining pattern (Fig. 1E). CD21 (1:50, clone 1F8, Dako) highlighted a follicular dendritic cell meshwork in the scattered germinal centers of reactive lymphoid follicles.
- Immunohistochemical staining for IgG (1:200, clone A57H, Dako) and IgG4 (1:200, clone HP6025, Invitrogen, Carlsbad, CA, USA) (Fig. 2) revealed a high proportion of plasma cells in the stroma. IgG4- and IgG-positive plasma cells were estimated by counting the most significantly infiltrative areas. More specifically, the number of IgG4- and IgG-expressing plasma cells in five high power fields (HPF) were counted, added together, and then divided by 5 to obtain the mean count. The IgG4/IgG ratio was also calculated from the mean count of IgG4 and IgG.
- Since the surgical excision, the patient has been in good general condition without a recurrent mass lesion in the breast for more than 10 months. Serum IgG and IgG4 levels were not evaluated before or after the excision. She has visited for follow-up, including a regular sonographic examination of her treated breast, every 6 months since the excision. No further treatments, such as steroid therapy, have been conducted, and no other masses have been detected.
CASE REPORT
- RDD was initially described as sinus histiocytosis with massive lymphadenopathy.1 It usually presents in children and young adults with bilateral cervical lymphadenopathy, and shows typical histologic features, including fibrosis, marked dilatation of sinuses with mixed inflammatory cells, and numerous histiocytic infiltration.9 In particular, infiltrating histiocytes have one large nucleus, conspicuous central nucleoli, and abundant clear and/or acidophilic cytoplasm. Emperiopolesis of lymphocytes, plasma cells, and red blood cells by large histiocytes is evident. Immunohistochemically, the histiocytes show reactivity for S-100 protein and CD68, which is helpful to verify the diagnosis of RDD. Extranodal manifestations can be present in any part of the body, including the breast. Compared to nodal RDD, extranodal RDD demonstrates more fibrosis, infrequent typical histiocytes, and a lesser degree of emperipolesis. To the best of our knowledge, 26 cases of RDD in the breast have been reported in the English literature,4 and the present case is the first report in Korea.
- Mass-forming lesions in breast, as manifestations of inflammatory infiltration, are occurred in variable diseases. With hitiocytic infiltration, several differential diagnoses can be considered, including Langerhans' cell histiocytosis, granulomatous lobular mastitis, extranodal RDD, and Erdheim-Chester disease, which are summarized in Table 1.
- Previously reported breast RDDs have included mammary manifestation of systemic RDD as well as primary RDD in mammary parenchyma and subcutis. In one case of mammary manifestation of systemic RDD, the patient had a history of RDD in the thigh and inguinal lymph node, and a mammary lesion consisting of numerous tiny nodules measuring up to 0.5 cm was found during routine screening.10 Primary RDDs of breast have been found in women of various ages, presenting as a painless, palpable mass for a few months duration.4 They were initially regarded as malignancies clinically and radiologically, and in all cases, excision or core needle biopsies were performed for diagnostic treatment. Among reported 26 cases, 3 patients had local recurrence ranging from 5 months to 2.5 years, and 1 died of disseminated RDD after 2 months post-operation. To date, all reported cases of RDD have shown characteristic histological findings. None of them exhibited IgG and/or IgG4 immunohistochemical staining for infiltrating plasma cells. Our case showed storiform sclerosing fibrosis with plasmacytic infiltration, rendering obliteration of preexisting normal breast lobules. These features somewhat overlap with IgG4-related sclerosing disease. More than 50 IgG4+ plasma cells/HPF or over 40% IgG4/IgG can be considered highly specific diagnostic criteria for IgG4-related sclerosing disease.3
- Recently, both secondary breast involvement of a systemic IgG4-related sclerosing disease and primary IgG4-related sclerosing mastitis have also been reported.3
- Five cases of primary IgG4-related sclerosing disease of the breast have been reported in the English literature.11,12 Ogiya et al.8 reported the first such case; the patient suffered from bilateral swelling of the breast and elevated serum IgG4, and was successfully treated by steroid therapy.11 Cheuk et al.11 subsequently described four cases of IgG4-related sclerosing disease in the breast, and proposed the term IgG4-related sclerosing mastitis.12 In that report, the patients presented with painless masses in the breast, and underwent surgical excision without recurrence. Compared with lymphocytic mastitis and granulomatous mastitis, IgG4-related mastitis shows denser IgG4+ plasmacytic infiltration (272-495/HPF) with higher IgG4/IgG ratios in the range of 49-85%.
- A possible link between RDD and IgG4-related sclerosing disease has been suggested because they have some overlapping histological features. In 2009, Kuo et al.10 described a cutaneous RDD with numerous IgG4-positive plasma cells, which was the first report of overlapping findings between the two diseases.5-7,13 Plasmacytic infiltration is a relatively consistent finding in RDD, and fibrosis is also common in extranodal cases. Our case also demonstrated features of IgG4-related sclerosing lesions such as fibrosis and many IgG4-positive plasma cells in the lesion in addition to the typical histological findings of RDD. In the present case, the mean count of IgG4-positive cells was 100.2/HPF, and the ratio of IgG4-positive plasma cells to IgG-positive plasma cells was 56.7%, which met the diagnostic criteria of IgG4-related sclerosing disease histologically. To the best of our knowledge, this is the first case of RDD in the breast associated with prominent IgG4-positive plasmacytic infiltration and fibrosis.
- Although the diagnostic criteria of IgG4-related sclerosing disease are established, diagnosis should be made carefully, considering clinical, laboratory, radiologic, and histological features. IgG4-related sclerosing disease is now considered an immunity matter, and steroid therapy is known to be effective. Some authors have suggested that steroid therapy, which is now considered the choice of treatment for IgG4-related sclerosing disease, could be a useful treatment modality for RDD with features of IgG4-related sclerosing disease.6 To clarify the pathogenic relationships between these two diseases, comprehensive studies that consider clinical, laboratory, radiologic, and histopathologic features are required.
DISCUSSION
- 1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 63-70. PubMed
- 2. Chen J, Tang H, Li B, Xiu Q. Rosai-Dorfman disease of multiple organs, including the epicardium: an unusual case with poor prognosis. Heart Lung 2011; 40: 168-171. ArticlePubMed
- 3. Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010; 17: 303-332. PubMed
- 4. Tenny SO, McGinness M, Zhang D, Damjanov I, Fan F. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J 2011; 17: 516-520. ArticlePubMed
- 5. Chen TD, Lee LY. Rosai-Dorfman disease presenting in the parotid gland with features of IgG4-related sclerosing disease. Arch Otolaryngol Head Neck Surg 2011; 137: 705-708. ArticlePubMed
- 6. Roberts SS, Attanoos RL. IgG4+ Rosai-Dorfman disease of the lung. Histopathology 2010; 56: 662-664. ArticlePubMed
- 7. Richter JT, Strange RG Jr, Fisher SI, Miller DV, Delvecchio DM. Extranodal Rosai-Dorfman disease presenting as a cardiac mass in an adult: report of a unique case and lack of relationship to IgG4-related sclerosing lesions. Hum Pathol 2010; 41: 297-301. ArticlePubMed
- 8. Ogiya A, Tanaka K, Tadokoro Y, et al. IgG4-related sclerosing disease of the breast successfully treated by steroid therapy. Breast Cancer 2010 11 03 [Epub]. http://dx.doi.org/10.1007/s12282-010-0225-6. ArticlePDF
- 9. Shrestha B, Sekiguchi H, Colby TV, et al. Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with autoimmune pancreatitis: report of 6 and 12 cases with similar histopathology. Am J Surg Pathol 2009; 33: 1450-1462. PubMed
- 10. Kuo TT, Chen TC, Lee LY, Lu PH. IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease: an additional immunohistochemical feature and possible relationship to IgG4-related sclerosing disease. J Cutan Pathol 2009; 36: 1069-1073. ArticlePubMed
- 11. Cheuk W, Chan AC, Lam WL, et al. IgG4-related sclerosing mastitis: description of a new member of the IgG4-related sclerosing diseases. Am J Surg Pathol 2009; 33: 1058-1064. ArticlePubMed
- 12. Rosai J. Rosai and Ackerman's surgical pathology. 2004; Edinburgh, New York: Mosby, 1911-1913.
- 13. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19-73. PubMed
REFERENCES
Fig. 1Mammography revealing an ill-defined mass in the right lower breast (A). Numerous histiocytes showing pink, abundant cytoplasm (B) containing intact inflammatory cells (emperipolesis, arrows) (C). Nuclear and cytoplasmic S-100 protein expression of histiocytes highlighting the entrapped inflammatory cells in their cytoplasm, which are negative for S-100 protein (D). Histiocytes expressing CD68 in cytoplasm (E).



Figure & Data
References
Citations
Citations to this article as recorded by 
- Extranodal Rosai–Dorfman Disease With Increased IgG4-Positive Plasma Cells Involving the Breast: A Case Report With Review of the Literature
Raymond Chimatira, Raisa Wessels
The American Journal of Dermatopathology.2026; 48(5): 391. CrossRef - From Tumor Suspicion to Immune Revelation: A Breast Lump Signals IgG4-related Disease
Renu Singh, Gurwinder Kaur, Md Ali Osama, Chitresh Kumar, Devender Bairwa
Indian Journal of Gynecologic Oncology.2026;[Epub] CrossRef - Extranodal Rosai–Dorfman Disease of the Male Breast: A Case Report and Literature Review
Raymond Chimatira, Dharshnee R. Chetty
The American Journal of Dermatopathology.2026; 48(6): 452. CrossRef - Rosai-Dorfman Disease of the Breast: Radiologic-Pathologic Correlation
Kathryn W Zamora, Stefanie Zalasin, Ami Desai, Hua Guo
Journal of Breast Imaging.2025; 7(5): 576. CrossRef - Diagnosis and management of Rosai–Dorfman disease of the breast: Case report and literature review
Jessman King Lun Lo, Sara Wai Wun Fung, Zara Chui San Tsang
Surgical Practice.2024; 28(4): 229. CrossRef - IgG4-related Breast Disease: Review of the Literature
Helana Jeries, Yolanda Braun-Moscovici, Alexandra Balbir-Gurman
Rambam Maimonides Medical Journal.2024; 15(4): e0018. CrossRef - Pathology of IgG4-related sclerosing mastitis
Polycarp Erivwo, Gulisa Turashvili
Journal of Clinical Pathology.2021; 74(8): 475. CrossRef - Rosai–Dorfman Disease: Breast Involvement—Case Report and Literature Review
George Iancu, Nicolae Gica, Laura Mihaela Mustata, Anca Maria Panaitescu, Danut Vasile, Gheorghe Peltecu
Medicina.2021; 57(11): 1167. CrossRef - Sonographic features of Rosai‐Dorfman disease in the breast: A case report
Gi W. Shin, Young M. Park, Young J. Heo, Jin W. Baek, Yoo J. Lee, Ji Y. Han, Hayoung Park
Journal of Clinical Ultrasound.2020; 48(2): 108. CrossRef - Rosai-Dorfman Disease of the Breast With Variable IgG4+ Plasma Cells
Jenny C. Hoffmann, Chieh-Yu Lin, Siddhartha Bhattacharyya, Olga K. Weinberg, Karen M. Chisholm, Michael Bayerl, Michael Cascio, Girish Venkataraman, Kimberly Allison, Megan Troxell, Chung-Che Chang, Adam Bagg, Tracy I. George, Dennis O’Malley, Robert S. O
American Journal of Surgical Pathology.2019; 43(12): 1653. CrossRef - Rosai-Dorfman disease presenting as a breast mass
Ding Dai, Qi Cai, Nasreen A. Vohra, Jan Wong, Zsuzsanna P. Therien, Karlene Hewan-Lowe, Ann Sutton
Archives of Pathology and Clinical Research.2019; 3(1): 008. CrossRef - Erdheim-Chester Disease Involving Lymph Nodes and Liver Clinically Mimicking Lymphoma: A Case Report
Yeoun Eun Sung, Yoon Seo Lee, Jieun Lee, Kyo Young Lee
Journal of Pathology and Translational Medicine.2018; 52(3): 183. CrossRef - Enfermedad de Rosai-Dorfman en mama de paciente masculino: una entidad rara
Paola Iturralde Rosas-Priego, José Daniel Flores-Alatriste, Daniela Stuht López, Javier Gómez Pedroso-Rea, Cecilia Ortiz-de-Iturbide, Jorge Valenzuela-Tamariz, Manuel Ubiergo-García
Revista de Senología y Patología Mamaria.2018; 31(2): 72. CrossRef - Increased Immunoglobulin G4-positive Plasma Cells in Lymphadenoma of the Salivary Gland: An Immunohistochemical Comparison Among Lymphoepithelial Lesions
Jiyoon Kim, Joon Seon Song, Jong-Lyel Roh, Seung-Ho Choi, Soon Yuhl Nam, Sang Yoon Kim, Kyung-Ja Cho
Applied Immunohistochemistry & Molecular Morphology.2018; 26(6): 420. CrossRef - IgG4-Related Sclerosing Disease of the Breast in a Male Patient
Taisia Vitkovski, Galina S. Marder, Dominic A. Filardi, Ekta Gupta, Frank Breuer
International Journal of Surgical Pathology.2017; 25(8): 711. CrossRef - Extranodal manifestation of Rosai-Dorfman disease in the breast tissue
Qiao Zhou, Umer Ansari, Nandan Keshav, Fiona Davis, Maria Cundiff
Radiology Case Reports.2016; 11(3): 125. CrossRef - Rosai-Dorfman Disease with Massive Cutaneous Nodule on the Shoulder and Back
Han Ma, Yue Zheng, Guoxing Zhu, Jie Wu, Chun Lu, Wei Lai
Annals of Dermatology.2015; 27(1): 71. CrossRef - IgG4-related disease of the breast: a systemic disease whose mammary manifestations mimic breast cancer
Takuya Moriya, Hisashi Hirakawa, Maki Nagashima, Mitsuhiko Yasuda, Izo Kimijima
International Cancer Conference Journal.2015; 4(2): 67. CrossRef - Primary Cutaneous Marginal IgG4 Lymphoma and Rosai–Dorfman's Disease Coexisting in Several Lesions of the Same Patient
Salma Machan, Camino Medina, Socorro María Rodríguez-Pinilla, José M. Suárez-Peñaranda, Yolanda Castro, Paula Molés, Celia Requena, Carles Saus, Luis Requena, Carlos Santonja
The American Journal of Dermatopathology.2015; 37(5): 413. CrossRef - A subset of Rosai–Dorfman disease cases show increased IgG4‐positive plasma cells: another red herring or a true association with IgG4‐related disease?
Madhu P Menon, Moses O Evbuomwan, Juan Rosai, Elaine S Jaffe, Stefania Pittaluga
Histopathology.2014; 64(3): 455. CrossRef - Freiburg Neuropathology Case Conference: A Partially Calcified, Dura-based Tumour of the Frontal Lobe
C. A. Taschner, O. Staszewski, R. Jabbarli, A. Keuler, M. Prinz
Clinical Neuroradiology.2013; 23(1): 63. CrossRef - Pulmonary IgG4+ Rosai-Dorfman disease
Karim El-Kersh, Rafael L Perez, Juan Guardiola
BMJ Case Reports.2013; 2013: bcr2012008324. CrossRef
 PubReader
PubReader Cite this Article
Cite this Article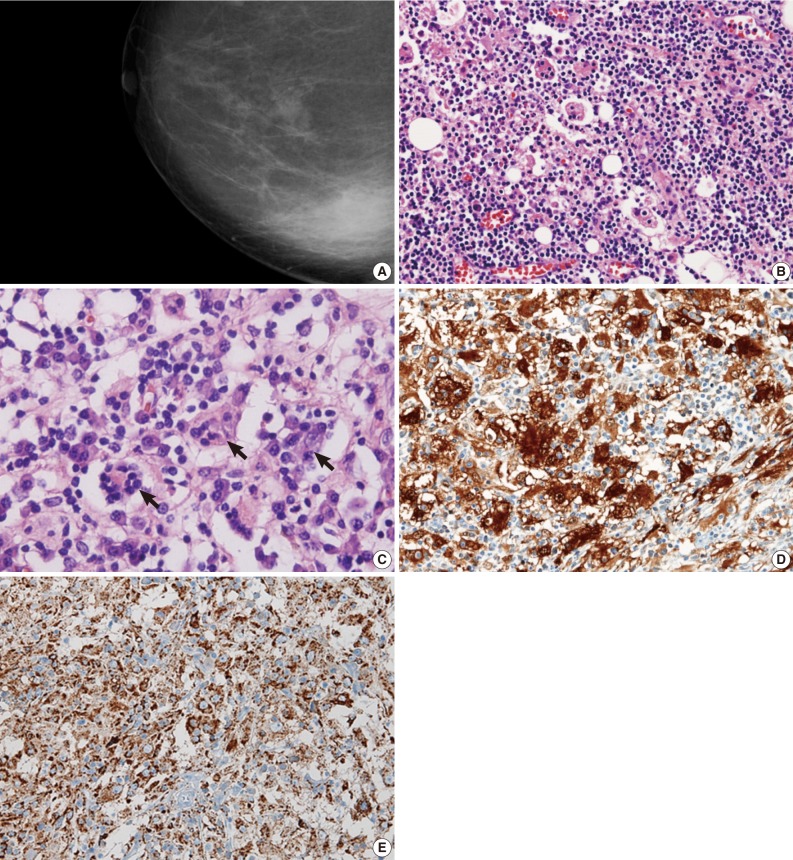

Rosai-Dorfman Disease in the Breast with Increased IgG4 Expressing Plasma Cells: A Case Report


Fig. 1 Mammography revealing an ill-defined mass in the right lower breast (A). Numerous histiocytes showing pink, abundant cytoplasm (B) containing intact inflammatory cells (emperipolesis, arrows) (C). Nuclear and cytoplasmic S-100 protein expression of histiocytes highlighting the entrapped inflammatory cells in their cytoplasm, which are negative for S-100 protein (D). Histiocytes expressing CD68 in cytoplasm (E).
Fig. 2 Normal breast lobules are not identifiable due to sclerotic, fibrous stroma (A). Many IgG-expressing plasma cells (B) also showing reactivity for IgG4 (C).
Fig. 1
Fig. 2
Rosai-Dorfman Disease in the Breast with Increased IgG4 Expressing Plasma Cells: A Case Report

Table 1 Differential diagnosis of non-epithelial mass forming lesion in breast with hitiocytes
RDD, Rosai-Dorfman disease; LCH, Langerhans' cell histiocytosis.

