 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 46(2); 2012 > Article
-
Original Article
Evaluation of Bronchiolar and Alveolar Cell Injuries Induced by Short- and Long-term Exposure to Sidestream Smoke - Kun-Young Kwon, Hye-Ra Jung, Ilseon Hwang, Won-Il Choi1
-
Korean Journal of Pathology 2012;46(2):151-161.
DOI: https://doi.org/10.4132/KoreanJPathol.2012.46.2.151
Published online: April 25, 2012
Department of Pathology, Keimyung University School of Medicine, Daegu, Korea.
1Department of Internal Medicine, Keimyung University School of Medicine, Daegu, Korea.
- Corresponding Author: Kun-Young Kwon, M.D. Department of Pathology, Keimyung University School of Medicine, 56 Dalseong-ro, Jung-gu, Daegu 700-712, Korea. Tel: +82-53-580-3812, Fax: +82-53-580-3823, k19156ky@dsmc.or.kr
© 2012 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- We investigated effects of short- and long-term exposure to sidestream smoke on the bronchiolar and alveolar cells in Sprague-Dawley rats.
-
Methods
- Rats were divided into five experimental groups: groups 1, 2, and 3 (1-month exposure to 3, 5, and 7 cigarettes a day, respectively), groups 4 and 5 (3- and 6 month exposure to five cigarettes a day, respectively). We examined the morphologic changes, the expressions of tumor necrosis factor α (TNF-α), tumor growth factor β1 (TGF-β1), interlekin (IL)-1α, IL-1β, Ki-67, and cytokeratin 14 and in situ apoptosis in the bronchiolar and alveolar cells on light microscopy (LM) and electron microscopic (EM) terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining.
-
Results
- LM showed the respiratory bronchiolar dilatation and alveolar wall collapse. In groups 3, 4, and 5, EM showed loss of the cilia and Clara cells with irregular size, more prominent alveolar wall collapse and dilation of alveolar duct than those of groups 1 and 2. Bronchiolar and alveolar cells showed increased expressions of TNF-α and TGF-β in groups 4 and 5. LM and EM TUNEL stains showed increased apoptosis in groups 3, 4, and 5.
-
Conclusions
- Sidestream smoke causes a bronchiolar and alveolar cell injury and the severity correlates strongly the volume and duration of exposure to sidestream smoke.
- Animal model
- Thirty-five male Sprague-Dawley rats weighing 200-250 g were used for this study. Animals were provided with commercially available rat feed and drinking water, and were subjected to a 1-week accommodation period prior to the experimental procedure. Rats were divided into two groups: the control group and the experimental group, each of which comprises five rats. Rats of the experimental group were exposed to sidestream smoke in smoking chambers that we have designed ourselves. We made a model of sidestream smoke using plastic chambers where experimental rats can inhale the sidestream smoke from the cigarette end rather than can direct aspirate the mainstream smoke of cigarettes after the cigarette was lighted. Rats of the control group were only provided with commercial rat feed and drinking water, but not exposed to the cigarette smoke. Rats of the experimental groups were exposed to the sidestream smoke as follows:
- Morphologic changes of the bronchioles and alveolar cells were examined on the LM and the EM after both short- and long-term exposure to cigarette smoke. Tissue specimens were prepared for the LM, transmission EM (TEM) and scanning EM (SEM) with the routine procedures. EM findings were analyzed semi-quantitatively and then assessed by a relative comparison with the control group based on the following criteria: minimal (+/-) (similar to slight change as compared with the control group), mild (+) (focal and mild change on EM at a magnification of ×10,000), moderate (++) (focal but distinct change on EM at a magnification of ×5,000) and prominent (+++) (diffuse and prominent change on EM at a magnification of ×5,000).
- Immunohistochemistry
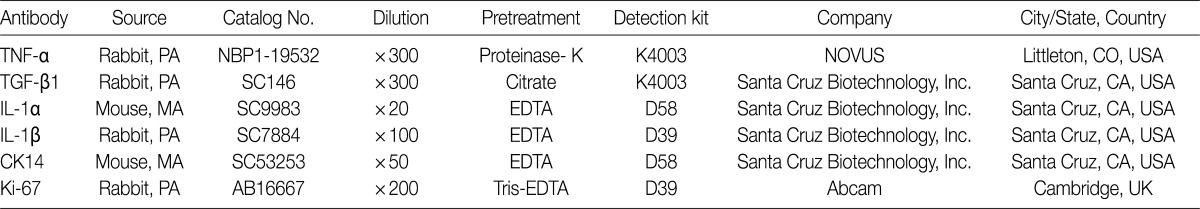
- We performed immunohistochemical stainings of TNF-α, TGF-β1, IL-1α, IL-1β, Ki-67, and CK14 in bronchioles and alveolar cells. Formalin-fixed, paraffin-embedded lung tissue sections (5-µm thick) were placed in a 60℃ oven for 30 minutes and then were deparaffinized in xylene three times for 20 minutes each. The deparaffinized sections were then rehydrated by passage through a graded series of ethanol to water. The sections were then heated for 25 minutes in citrate-buffered saline (pH 6.0) for antigen retrieval. Endogenous peroxidases were blocked with a solution of 3% H2O2 in water for ten minutes. The tissue sections were incubated with primary antibodies against TNF-α, TGF-β1, IL-1α, IL-1β, CK14, and Ki-67 (Table 1). Diaminobenzidine (DAB, Dako, Carpinteria, CA, USA) was used as the chromogen. In addition, sections were counterstained with Mayer's hematoxylin. We analyzed the immunohistochemical expressions of available slides for interpretation in each group, and evaluated using a 5-point scoring system based on the percentage of cells expressing antigen with 0, ≤5%; 1, 6-20%; 2, 21-40%; 3, 41-70%; and 4, >70% where 5% was set at the cut-off value for positivity. The CK14 and Ki-67 antigens are expressed in the nuclei of bronchiolar basal cells and alveolar epithelial cells.
- In situ TUNEL staining
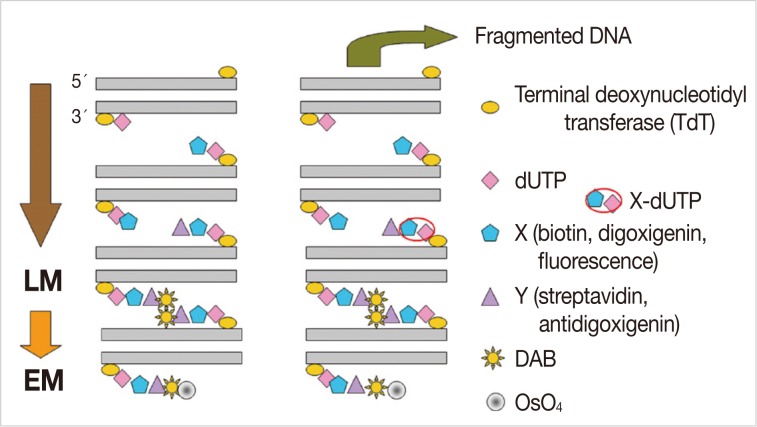
- We examined apoptosis in bronchiolar and alveolar cells on the LM and EM to detect the DNA fragments by the TUNEL technique. This method is based on the ability of terminal deoxynucleotidyl transferase (TdT) to catalyze the template-independent addition of biotinylated dUTP to the 3'-OH ends of double- or single-stranded DNA. In addition, we used the ApopTag peroxidase in situ apoptosis detection kit (Intergen, New York, NY, USA) to detect apoptosis in situ.
- LM TUNEL staining
- Paraffin-embedded tissue sections were cut into sections 5-µm thick, mounted on silane-coated glass slides and incubated for an hour at 60℃. The slides were deparaffinized in xylene three times for five minutes each, and then rehydrated through a graded alcohol series (100%, 95%, 70%, and 50%) for five minutes each. After washing with 0.01 M phosphate buffered saline (PBS) for five minutes, we digested the sections with proteinase K (20 µg/mL) at room temperature for 20 minutes, and washed them twice with distilled water for two minutes each. Endogenous peroxidase activity was blocked with 3% H2O2 in PBS for five minutes. The slides were rinsed twice with PBS for five minutes. For positive controls, sections were treated with 3% H2O2 and reacted with DNase I (1 or 2 U) at 37℃ for 30 minutes. Then, the sections were washed twice with PBS. The slides were covered with TdT buffer 75 µL and TdT/dUTP 50 µL and then left undisturbed for ten minutes. The sections were covered with plastic cover slips and incubated in a humidified chamber at 37℃ for 1.5 hours. For negative controls, sections were covered with reaction buffer alone and incubated under the same conditions. The slides were dipped into Coplin jars containing the stop buffer, agitated for 15 seconds and incubated for ten minutes at 37℃. Then the slides washed three times with PBS for five minutes each. The sections were covered with a peroxidase-coupled anti-digoxigenin antibody and incubated for 30 minutes at 37℃ in a humidified chamber. The slides were washed three times with PBS for two minutes each, and covered with 100 µL of DAB solution. The sections were counterstained with hematoxylin for one minute, washed with tap water, and dehydrated with a graded ethanol series. The sections were cleared with xylene and mounted with Permount. Five high-power fields were randomly selected for each slide. Non-parenchymal cells, including interstitial cells, were not analyzed. The total number of alveolar cells, TUNEL-positive alveolar cells, total bronchiolar epithelial cells and TUNEL-positive bronchiolar epithelial cells were counted. The percentage of TUNEL-positive cells was calculated for each slide, of which mean values were obtained for each experimental group.
- EM TUNEL staining
- The lung tissues were removed from rats of the experimental groups, sliced into small pieces (4×4×1 mm), and were fixed in periodate lysine paraformaldehyde solution at 4℃ overnight. After washing with 0.01 M PBS, we immersed the lung tissues in graded concentrations of sucrose in PBS (10% for an hour, 15% for two hours, and 20% for four hours) at 4℃. The tissues were then embedded in optimal cutting temperature (OCT) compound and frozen in a dry ice/acetone bath. The OCT-embedded tissue blocks were stored at -80℃, and then cut into 6 µm sections in a cryostat and mounted on silane-coated glass slides. The sections were air-dried and stored at -20℃ until for further laboratory procedure. After washing in 0.01 M PBS for five minutes, we incubated the sections with proteinase K (20 µg/mL) at room temperature for 15 minutes, and then washed them with distilled water (DW) five times, for two minutes each. Endogenous peroxidase activity was blocked with 3% H2O2 in methanol for 25 minutes. The slides were rinsed five times with DW for two minutes each, and incubated with 50 µL TdT reaction solution for three hours. The reaction was stopped by addition of 2× saline-sodium citrate for 15 minutes at room temperature. The sections were washed five times with 0.01 M PBS for two minutes each and then incubated with blocking solution containing 1% bovine serum albumin in PBS at room temperature for 20 minutes. Then the sections were treated with peroxidase-conjugated streptavidin for an hour at 37℃. The slides were washed in six changes of 0.01 M PBS for five minutes each. The sections were reacted with Tris-HCl solution for five minutes. Then, a 100 µL of DAB solution was added to each section. This was followed by the incubation of slides in the dark at room temperature for 3-5 minutes. The slides were placed in a Coplin jar and washed in 0.01 M PBS five times for two minutes each. For LM TUNEL staining, the sections were counterstained lightly with hematoxylin for one minute and washed with tap water. Following this, paraffin-embedded tissue samples were prepared in a stepwise manner. For EM TUNEL staining, the sections were reacted with 1% OsO4 in 0.01 M cacodylate buffer for an hour in a humidified chamber, and washed in three changes of 0.01M PBS for five minutes each. Then, the sections were dehydrated completely in a graded ethanol series, and embedded in inverted polyethylene embedding capsules filled with PolyBed mix resin (Poly/Bed 812, Polysciences Inc., Warrington, PA, USA). This was followed by a 3-day polymerization at 60℃. The gelatin capsules were removed from the glass slide, which was followed by the LM examination. Then we selected a portion for the EM examination. After trimming the PolyBed mix resin blocks, we performed ultrathin sections (100 nm) and examined the grids on a Hitachi H-7100 electron microscope (Tokyo, Japan) at 75 kV without routine double stains of uranyl acetate and lead citrate (Fig. 1).
- Western blot analysis of proform-PARP in apoptotic cells
- Lung tissues were homogenized in cell lysis buffer (0.05 M Tris, 0.15 M NaCl, 0.05 M ethylenediaminetetraacetic acid, 0.5% NP-40, 1 M dithiothreitol, 10 µg leupeptin, and 10 µg aprotinin; all from Sigma, St. Louis, MO, USA). The homogenate was incubated for 30 minutes on ice and centrifuged at 12,000 ×g for 30 minutes. The protein content of the supernatant was determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA). Samples were denatured by boiling with 4× sodium dodecyl sulfate (SDS) sample buffer for ten minutes. Proteins (50 µg per lane) were resolved by electrophoresis on an 8-12% SDS-polyacrylamide gel electrophoresis gradient gel at 100 V for three hours and transferred to nitrocellulose membrane (Millipore Co., Bedford, MA, USA). The membrane was further incubated with primary antibodies such as a polyclonal rabbit anti-PARP antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and β-actin (Santa Cruz Biotechnology). The membrane was subsequently incubated with appropriate secondary antibodies conjugated to horseradish peroxidase and developed in the ECL western detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
MATERIALS AND METHODS
- Gross and microscopic features
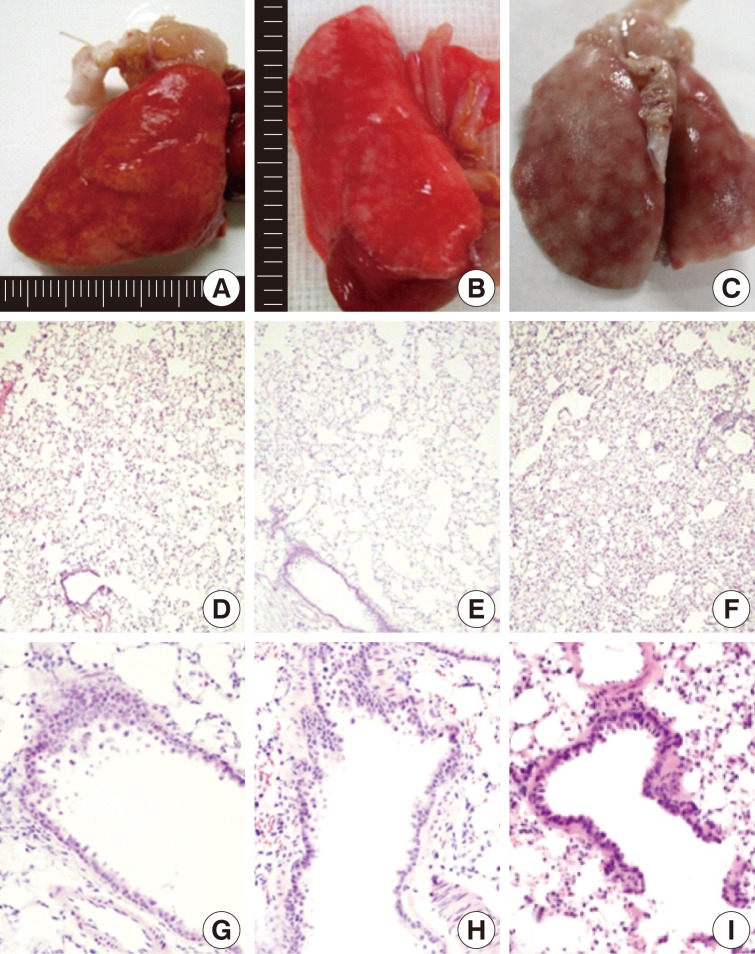
- On gross examination, there were mild patchy discoloration and congestion on the lung surface in rats of groups 1 and 2. In group 3, the lungs showed multifocal discoloration and congestion. In rats of groups 4 and 5, however, there were a patchy pale brown discoloration and a centrilobular patchy consolidation (Fig. 2A-C).
- Correlation of bronchiolar and alveolar cell changes with the volume of smoke exposure
- After a 1-month exposure to sidestream smoke, rats of group 1 showed a mild bronchiolar luminal dilatation, an irregular arrangement of the bronchiolar epithelial cell cilia and a patchy peribronchiolar inflammatory infiltrates. In rats of groups 2 and 3, there were more distinct changes in the bronchiolar cells as compared with the control group. In rats of groups 1, 2, and 3, there were patchy inflammatory infiltrates in the alveolar walls and air spaces around the respiratory bronchioles. But there were no notable differences in these findings between the three groups. In rats of group 2 and 3, there were a focal alveolar collapse and patchy emphysematous changes in the lung parenchyma (Fig. 2D-F).
- Correlation of bronchiolar and alveolar cell changes with the duration of smoke exposure
- As shown in Fig. 2G-I, there were morphological changes in the bronchiolar and alveolar cells after a 1-month exposure to sidestream smoke in group 2, after a 3-month exposure in group 4 and after a 6-month exposure in group 5. In rats of group 2, there was a bronchiolar dilatation with a mild proliferation of the bronchiolar epithelial cells accompanied by a focal, alveolar emphysematous change. In rats of group 4, there was a mild proliferation of the bronchiolar epithelial cells and this was accompanied by a peribronchiolar mild inflammatory infiltrate and a focal emphysematous change. In rats of group 5, there were a mild peribronchiolar thickening due to inflammation, multiple patchy areas of alveolar collapse and an emphysematous change.
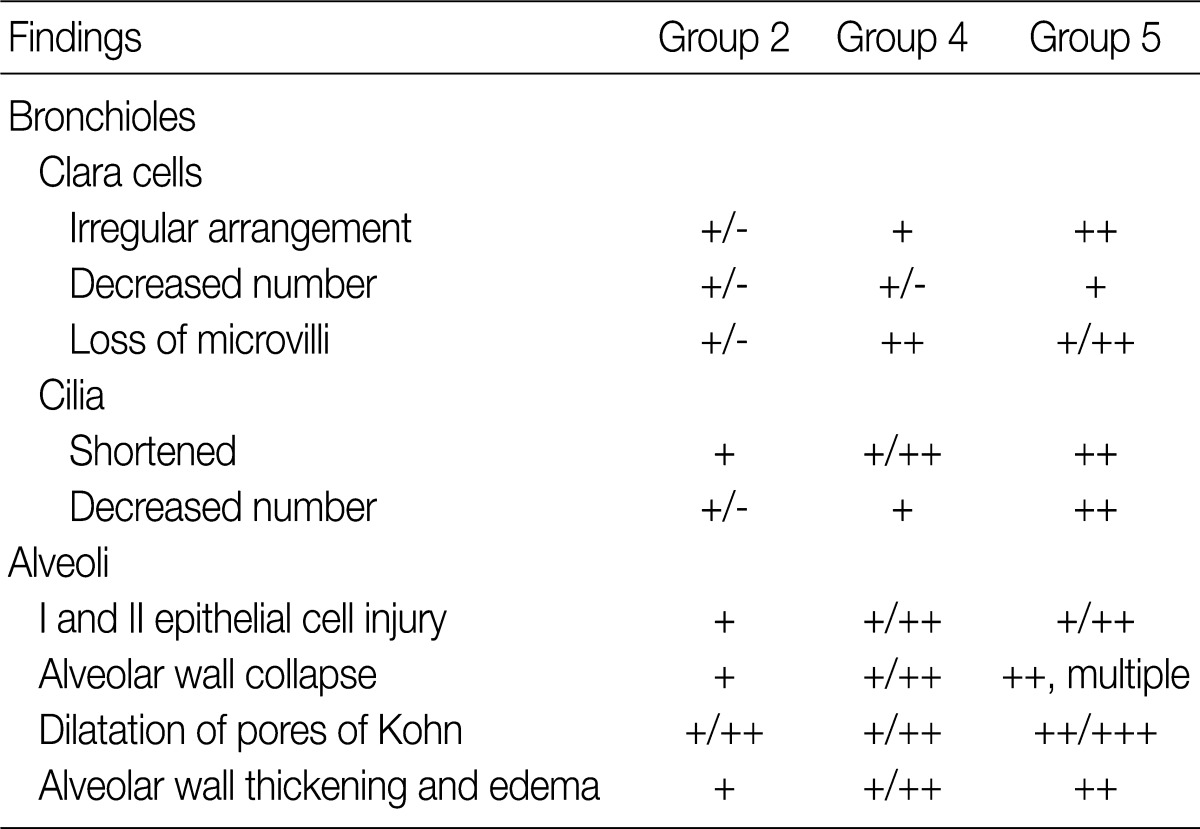
- Ultrastructural changes in the cilia of the bronchiolar epithelial cells, Clara cells, alveolar epithelial cells, and alveolar endothelial cells
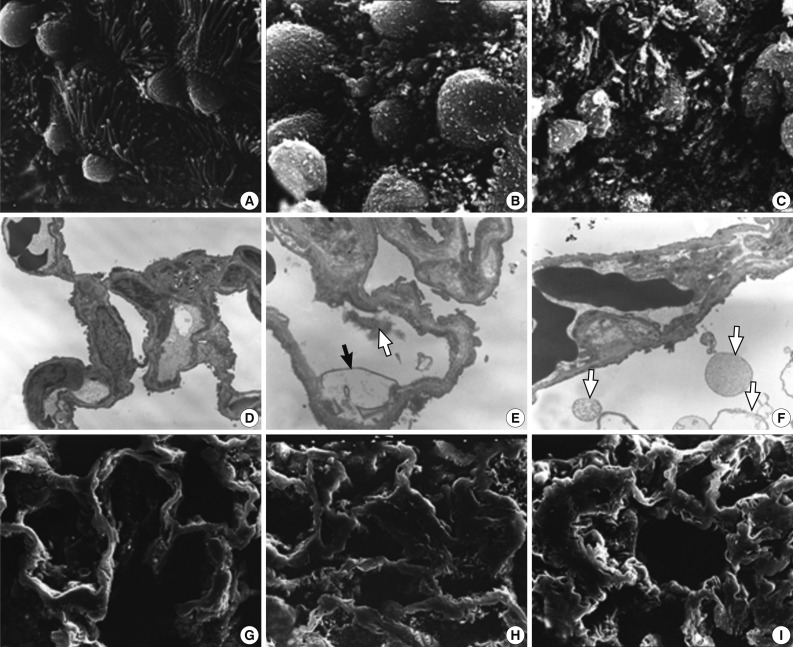
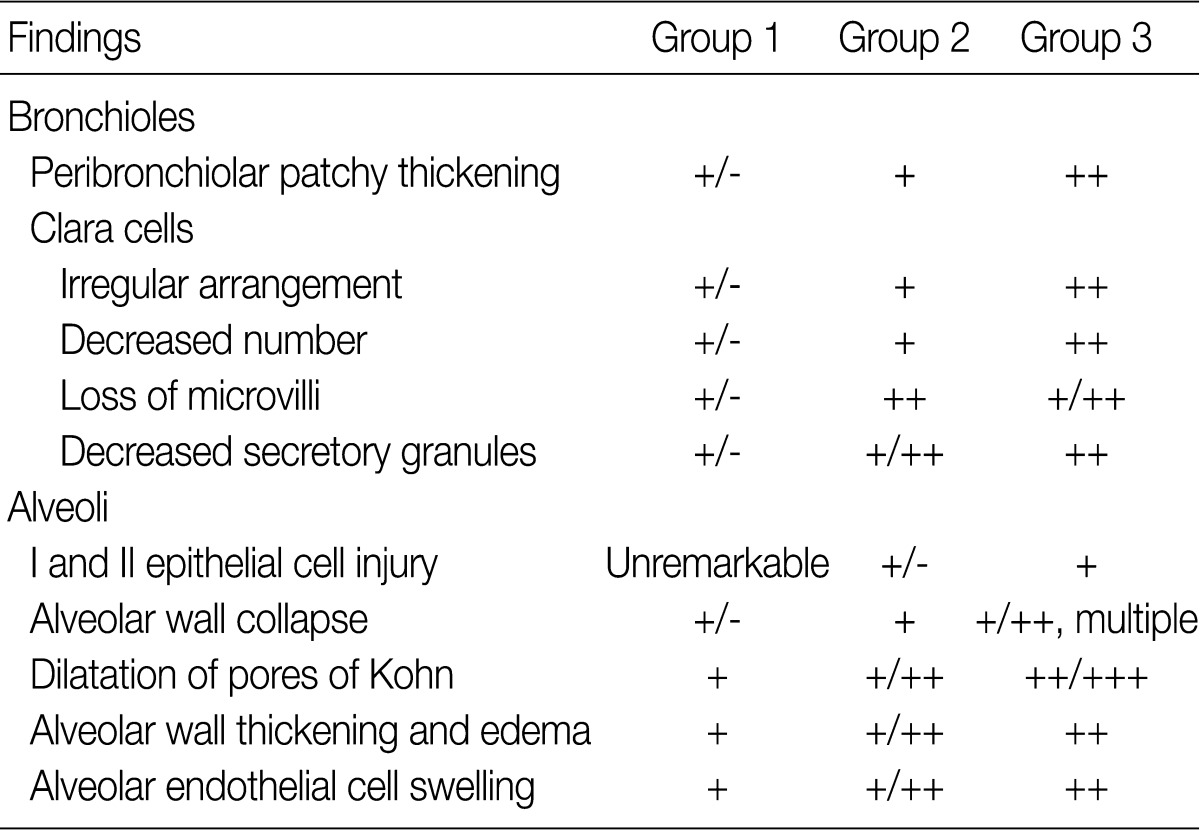
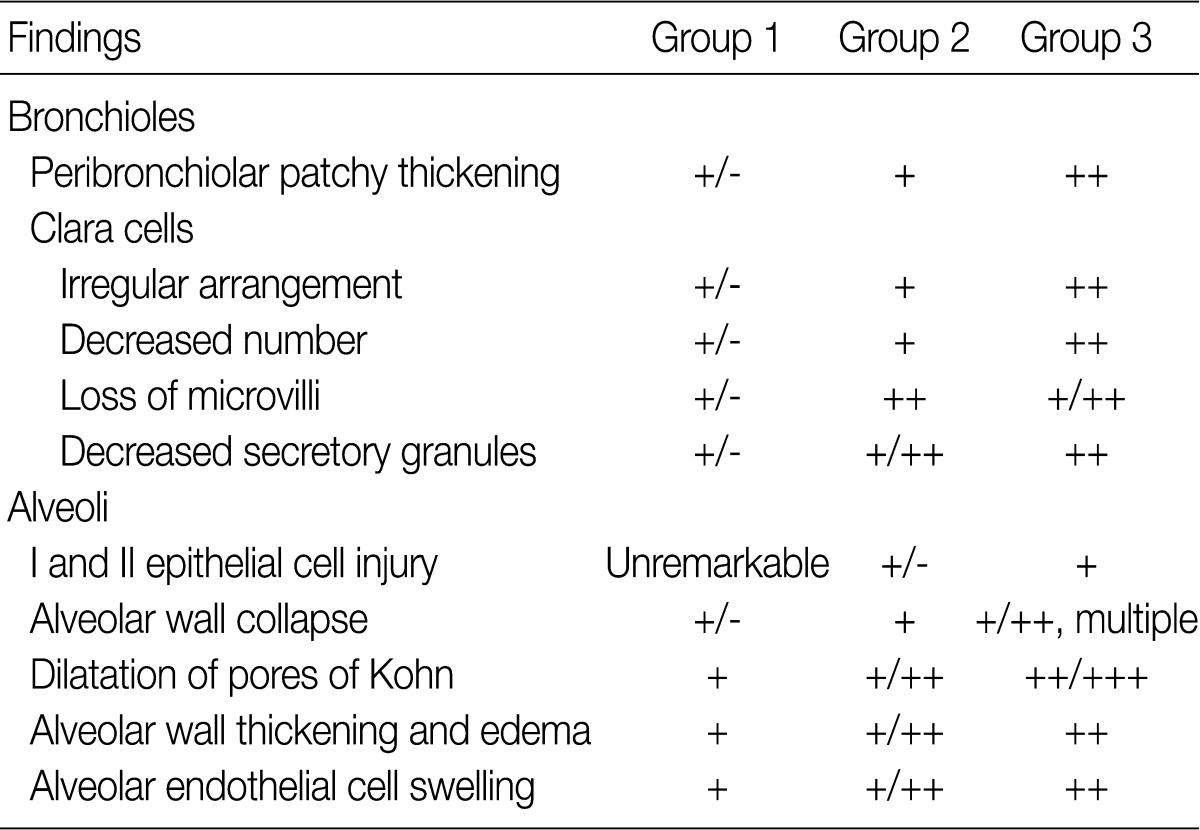
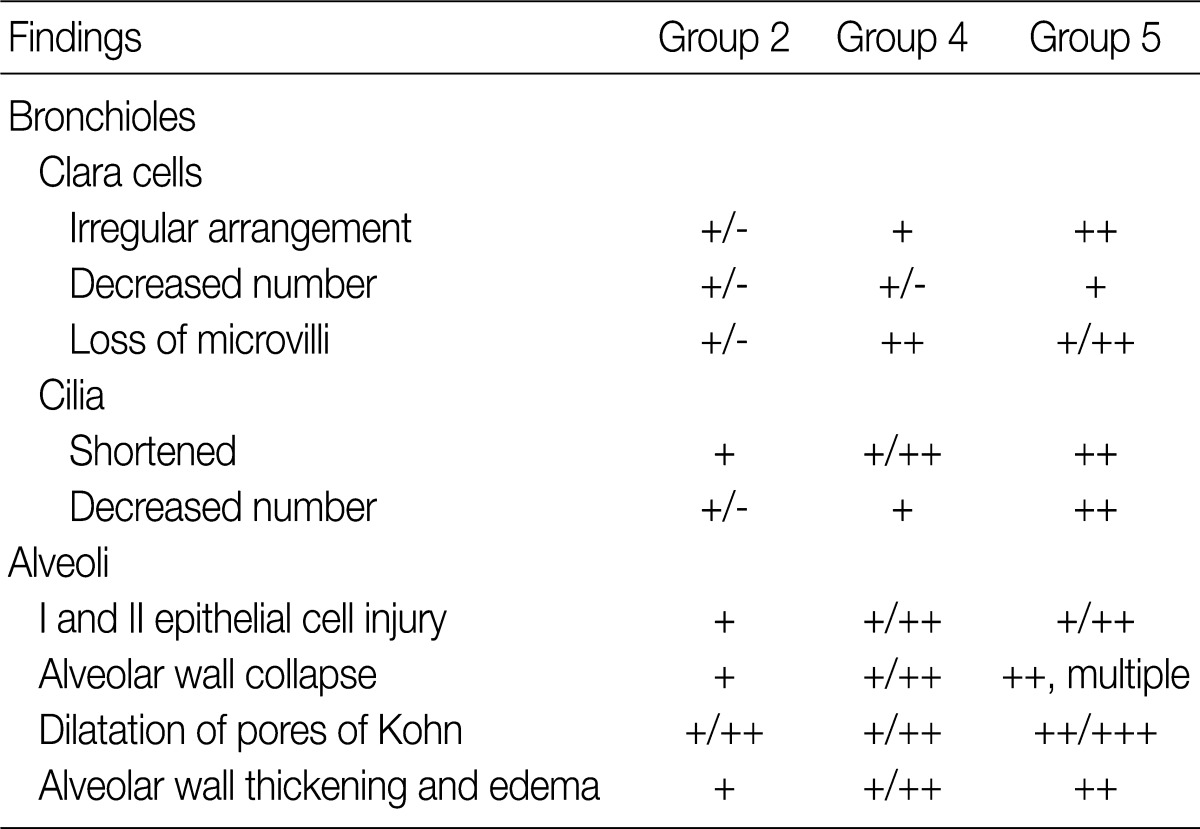
- The ultrastructural changes following an exposure to the smoke in the cilia of the bronchiolar epithelial cells, Clara cells, and alveolar epithelial cells are summarized in Tables 2 and 3. There were fewer terminal and respiratory bronchial mucosal cells, which was accompanied by the shortening or loss of the cilia of the epithelial cells. Clara cells were reduced in number, which showed an irregular distribution and a loss of surface microvilli. These findings were more prominent in rats of groups 4 and 5 which were exposed to a long-term sidestream smoke as compared with those of groups 1 to 3 which were exposed to a short-term one (Fig. 3A-C).
- In rats of groups 2 and 3, on the TEM examination of the lung parenchyma, there were an alveolar wall collapse with type I and type II epithelial cell injury, cytoplasmic bleb formation, free-floating cellular debris and mild endothelial cell swelling (Fig. 3D-F). In addition, on the SEM examination, there were a dilatation of the respiratory bronchiole and alveolar duct lumina, irregular dilatation of the pores of Kohn and the alveolar wall collapse and thickening. The SEM findings were more prominent in rats of group 3 as compared with those of group 1 (Fig. 3G-I). In rats of groups 4 and 5, there was an irregular dilatation of the alveolar structures and this was accompanied by the wall thickening and a focal emphysematous change.
- Immunohistochemical expression of cytokines and protein molecules
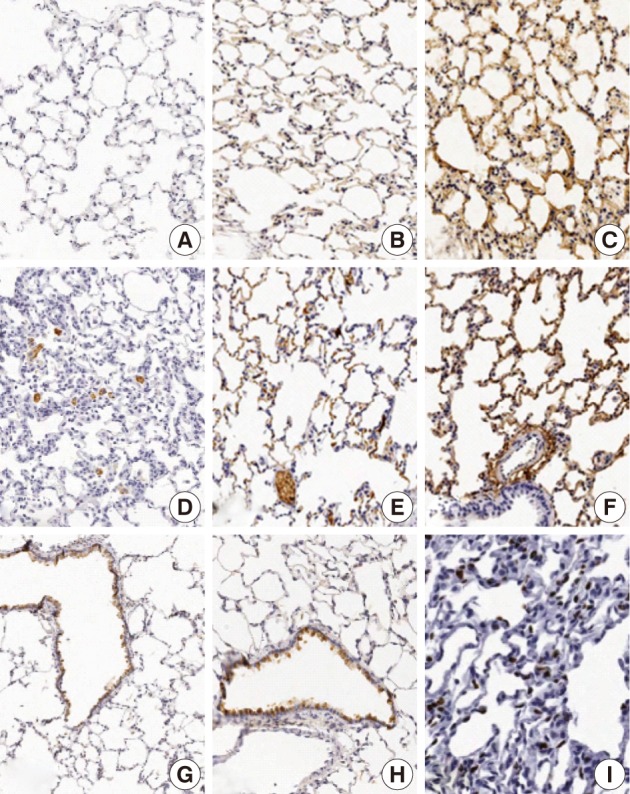
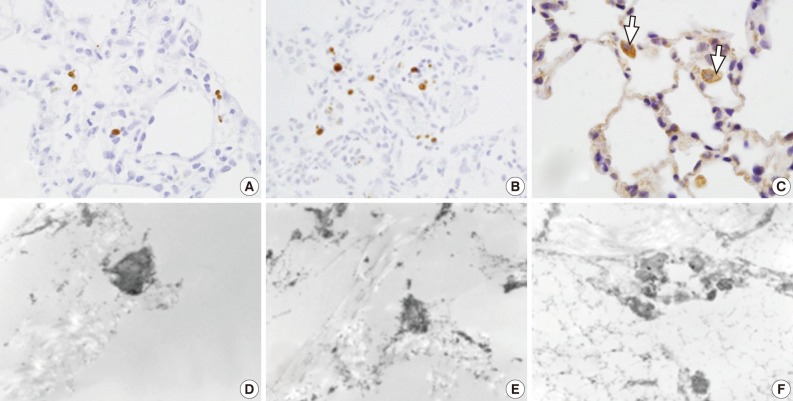
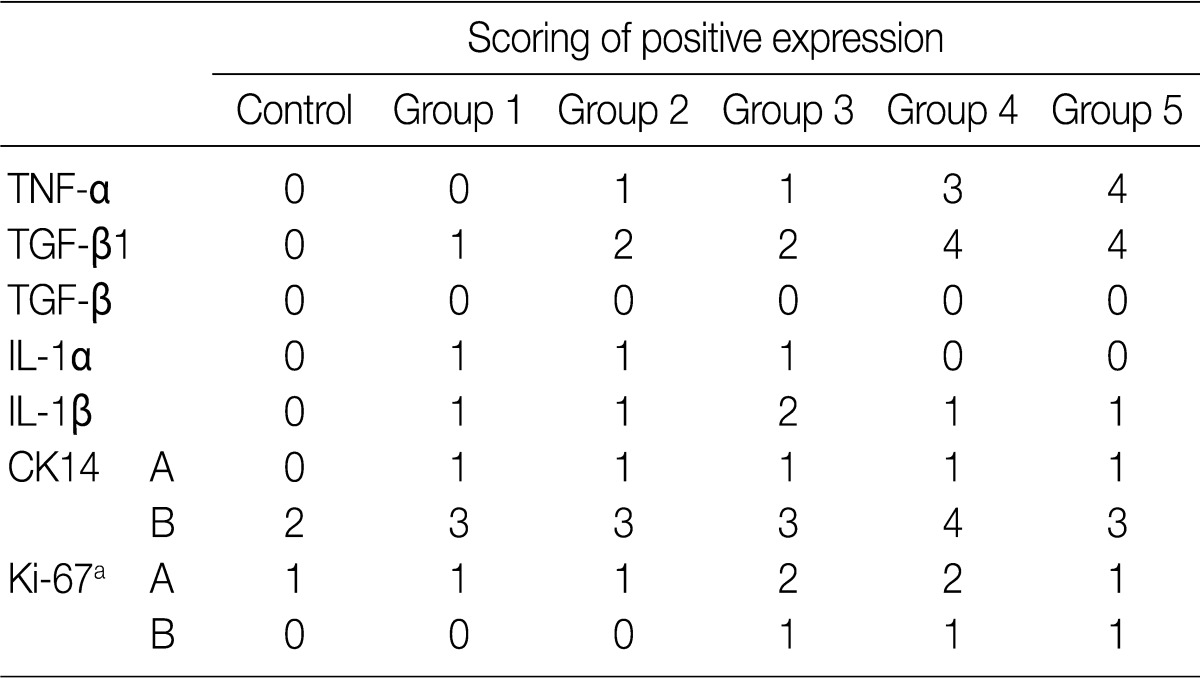
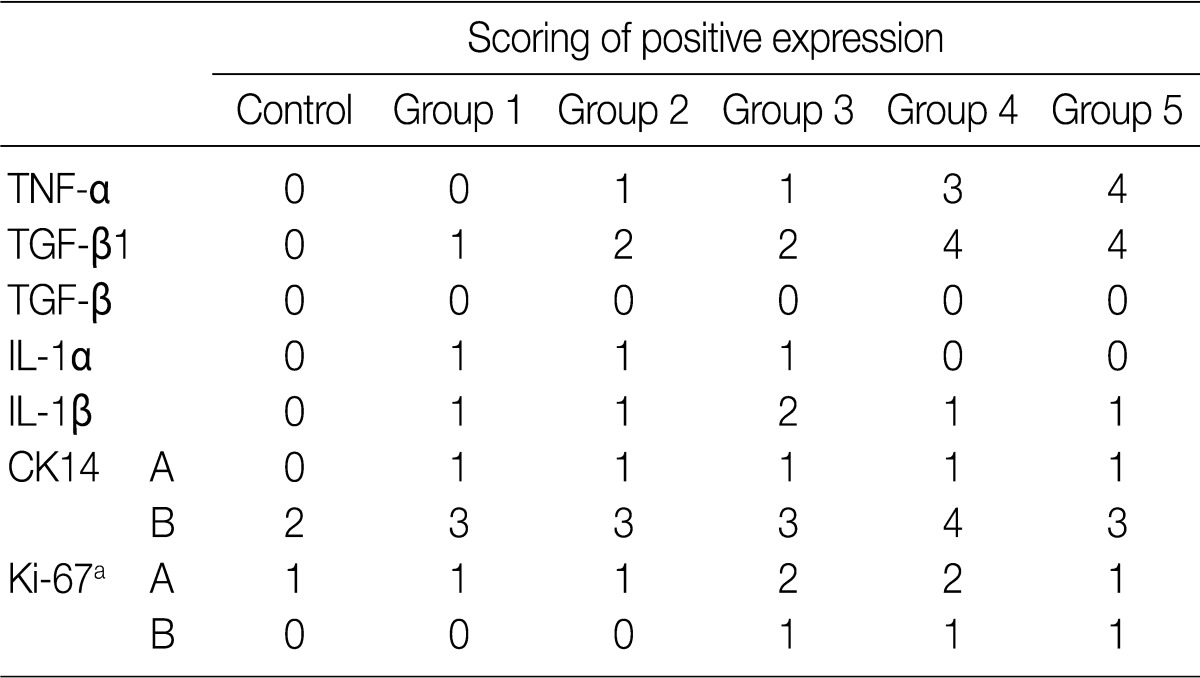
- The focal expression of TNF-α was found in the alveolar epithelial cells of rats of groups 2 and 3 (score, 1). Besides, there were the multiple patchy expression in group 4 (score, 3) and the diffuse expression along the alveolar epithelial cells in group 5 (score, 4) (Fig. 4A-C). There were no expression in rats of groups 1 and 3 and the weak expression of TNF-α in the bronchiolar epithelial cells of those of groups 4 and 5. The expression of TGF-β1 was found in the alveolar macrophages but was not seen in the alveolar cells of rats of group 1. In rats of groups 2 and 3, there was a multifocal expression (score, 2) of TGF-β1 in the alveolar endothelial and epithelial cells. In rats of groups 4 and 5, there was a diffuse expression of TGF-β1 (score, 4) in the alveolar endothelial and epithelial cells (Fig. 4D-F). The expression of TGF-β1 is not generally seen in the bronchiolar epithelial cells and Clara cells. The degree of the expression of IL-1α was not notable in the experimental groups. There were positive findings for the expression of IL-1β in the alveolar macrophages of groups 2, 3, 4, and 5, there was a diffuse expression of CK14 (score, 3 and 4) mainly in the bronchiolar epithelial cells and basal cells and a focal expression in the alveolar cells. However, the bronchiolar expression of CK14 did not vary among the groups (Fig. 4G, H). There was only a focal expression of Ki-67 (score, 1) in the bronchiolar cells of groups 3, 4, and 5 (Fig. 4I) (Table 4).
- Evaluation of TUNEL staining in bronchiolar and alveolar cells
- In rats of groups 1 and 2, on the LM TUNEL staining, there was evidence demonstrating that a few alveolar epithelial and endothelial cells, as well as alveolar macrophages, were scattered to undergo apoptosis. In the alveolar cells of groups 3, 4, and 5, the number of apoptotic cells was increased. The alveolar macrophages with cytoplasmic TUNEL staining were observed in rats of groups 4 and 5. But, there was no significant difference in the number of TUNEL-positive cells between groups 3, 4, and 5 (Fig. 5A-C). On the EM TUNEL staining, there were moderately electron dense apoptotic nuclei in the bronchiolar cells, alveolar epithelial cells, alveolar capillary endothelial cells, and in some scattered interstitial cells. On the EM TUNEL staining, there was also cytoplasmic expression in the alveolar macrophages. Apoptotic nuclei were occasionally seen in rats of group 2 but were frequently seen in those of groups 3 and 4 (Fig. 5D-F).
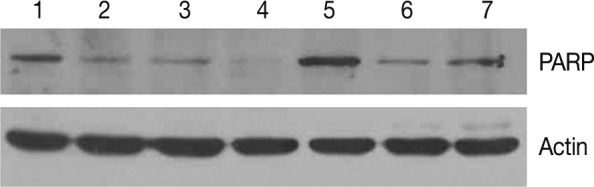
- Decreased expression of proform-PARP in apoptotic cells
- We examined the protein expression of anti-PARP on the western blot analysis. This showed that the expression of proform-PARP was decreased in groups 1, 2, and 3. Besides, there was a correlation between the decreased expression of proform-PARP and the volume of smoke exposure. In groups 4 and 5 where the experimental rats were exposed to a long-term sidestream smoke, the degree of the expression of proform-PARP was lower as compared with the control group (Fig. 6).
RESULTS
- COPD is defined as chronic inflammation of the airways and progressive destruction of lung parenchyma.2 Its pathogenesis is characterized by the abnormal enlargement of airspaces of the lung accompanied by destruction of the walls.14 Cigarette smoking is the main etiologic factor in COPD, of which emphysema and small airway disease are important pathologic manifestations.15 Oxidative stress caused by cigarette smoking is thought to induce a chronic inflammatory response in the lungs, which destroys the alveolar walls and induces emphysema. The alveolar cell damage and loss are caused by a mechanism that involves inhibition of cell proliferation and/or induction of cell death (apoptosis).16
- Sidestream smoke originates directly from the cigarette, and it has the same deleterious health effects as direct smoke. This causes both a medical and environmental concern.6,7 Sidestream smoke is associated with bronchial asthma, sudden infant death syndrome, chronic lung disease, lung cancer, ischemic heart disease and many other health problems.7-9,17 Despite the documented adverse effects of sidestream smoke on the lungs, only a limited number of objective methods and studies have analyzed the airway damage due to sidestream smoke. Similarly, there are few in vivo studies on the effects of passive smoking on the lung parenchyma and small airway.12,13 To date, however, no studies have reported the cytokine and protein molecular changes are associated with alveolar and bronchiolar cell injuries following an exposure to sidestream smoke. In the current study, we observed morphologic changes in bronchiolar and alveolar cells following an exposure to sidestream smoke. In addition, these findings were correlated the degree of damage with the duration and volume of an exposure to sidestream smoke in a vivo setting. We also analyzed the expression of TNF-α, TGF-β1, IL-1α, IL-1β, CK14, and Ki-67 in the current study.
- Microscopically, the more prominent morphological changes in the respiratory bronchioles and alveolar structure were present in the animals that were exposed to greater quantities of smoke and for longer duration. The animals exposed to smoke for only one month showed mainly dilatation of the respiratory bronchioles and alveolar ducts, with patchy alveolar wall collapse. In contrast, the animals exposed to smoke for three and six months showed a peribronchiolar patchy inflammatory thickening, as well as irregularity and expansion of the alveolar ducts and alveolar spaces. Such microscopic changes were similar to our previous findings with passive smoking.13 Our results therefore suggest that sidestream smoke induces remodeling of the respiratory bronchioles and alveolar parenchymal structure and this leads to early phase bronchiolocentric emphysematous change.
- There were characteristic ultrastructural findings in the animals exposed to sidestream smoke. The bronchioles showed an epithelial cell injury with an irregular arrangement, which was accompanied by the shortening and loss of cilia and the abnormalities of Clara cells. In the Clara cells whose number was reduced, there were an irregular distribution and a lack of the surface microvilli. As we have expected, our results showed that these bronchiolar changes were more prominent in the animals exposed to the sidestream smoke for longer periods. The alveolar cell damage included alveolar wall collapse with type I and type II epithelial cell injuries, endothelial cell swelling, and dilatation of the alveolar ducts and pores of Kohn. In the animals exposed to smoke for one month, these alveolar injuries were more prominent as the volume of smoke was increased. Rats exposed to smoke for longer times showed irregularly patchy dilatation of the alveolar spaces with wall thickening and destruction. Damages to the bronchiolar cilia and Clara cells were identified on the ultrastructural examination, which may adversely affect the defense mechanism in the lungs and then lead to development of small airway and lung parenchymal diseases.13,18
- On immunohistochemistry, little or no expression of TNF-α was detected in rats of groups 1, 2, and 3. In rats of group 4, the multifocal patchy expression was increased. In rats of group 5, there was a diffuse expression along the alveolar walls. Watanabe et al.19 reported that smoke has pro-inflammatory effects and stimulates TNF-α release by alveolar macrophages and possibly by pulmonary alveolar cells. The smoke-induced activation of TNF-α secretion usually requires a significant amount of smoke exposure.19 This is consistent with the reports made by Zoppini et al.20 who showed markedly increased levels of TNF-α in response to increased or long-term cigarette smoking. As a pro-inflammatory cytokine, TNF-α is believed to play a central role in the induction and maintenance of airway inflammation in COPD.21 Zhang et al.15 reported that alveolar cell damage mediated by TNF-α may have an important role in the development of emphysema, and TNF inhibitors such as rhTNFR:Fc can reduce the level of alveolar cell apoptosis in smoking rats.
- In the current study, there was a multifocal expression of TGF-β1 (40%) in groups 2 and 3 where the experimental rats were subjected to a short-term exposure to sidestream smoke. In addition, in groups 4 and 5 where the experimental rats were subjected to a long-term exposure to sidestream smoke, there was a more diffuse expression of TGF-β1 (80%) in the alveolar endothelial and epithelial cells. The expression of TGF-β1 is generally seen in the bronchiolar epithelial cells and Clara cells. TGF-β1 inhibits cell proliferation by inducing growth arrest in the G1 phase of cell cycle and regulating the activities of several inhibitors of cyclin-dependent kinases (CDKs).22 In addition, TGF-β1 induces oxidative stress that may regulate cell proliferation and the cell cycle.22 TGF-β1 gene expression is increased in the alveolar walls of smokers, particularly those who develop COPD.22 Marwick et al.16 reported that cigarette smoke-induced oxidative stress and TGF-β1 may inhibit cellular proliferation by the CDK inhibitor p21Waf1/Cip1 in human alveolar epithelial type II cells, and this may be relevant to the loss of alveolar cells in emphysema. Despite the known effects of direct tobacco smoke on the expressions of TNF-α and TGF-β1, no previous in vivo studies have shown that long-term sidestream smoking can induce activation of TNF-α and TGF-β1 in pulmonary alveolar and bronchiolar epithelial cells.
- Our results also showed that there was a focal expression of CK14 (40%) in the bronchiolar epithelial cells but no expression in the alveolar cells of the control rats. In the experimental rats which were not exposed to sidestream smoke, there were a multifocal expression of CK14 in the alveolar epithelial cells (15-20%) and a diffuse expression in the bronchiolar epithelial and basal cells (60-80%). Singh et al.23 reported that CK14 selectively labeled the basal cells of the normal esophageal epithelium and all the esophageal squamous carcinoma cells and adjacent carcinoma in situ. Xue et al.24 reported that CK14 was expressed in the mild to moderate dysplastic areas, and was diffusely expressed in the areas of carcinoma in situ and squamous cell carcinoma. In the current study, there were no dysplastic changes in the proliferated bronchiolar epithelial cells. Lee et al.25 have reported not only that the Ki-67 labeling index is strongly associated with heavy smoking or a long-term exposure to smoke but also that it is abnormally higher in the bronchioles of smokers despite a lack of the squamous metaplasia or dysplasia. Our results showed that the Ki-67 labeling index was slightly increased in the alveolar cells in the experimental groups (20-30%) compared to the control group (10%). But there was no significant difference in the degree of the expression of Ki-67 among the experimental groups.
- In rats of groups 1 and 2, on the LM TUNEL staining, there was evidence demonstrating that a few alveolar epithelial and endothelial cells, as well as alveolar macrophages, were scattered to undergo apoptosis. In the alveolar cells of groups 3, 4, and 5, apoptotic nuclei were seen in the bronchiolar and alveolar epithelial cells. In groups 4 and 5, the LM TUNEL stain also revealed cytoplasmic expression of the alveolar macrophages, suggesting an active phagocytosis of the apoptotic nuclear debris. Moreover, on the EM TUNEL staining, there were moderately electron dense apoptotic nuclei in groups 2, 3, and 4. The apoptotic nuclei were more easily seen on EM TUNEL staining. This clarified the equivocal appearances of TUNEL-positive cells in rats of group 2 on the LM. On the EM TUNEL staining, the early phase of apoptosis was also observed. Chronic exposure of rats to mainstream cigarette smoke induces a significant and time-dependent increase in the appearance of apoptotic cells in the bronchial, bronchiolar epithelium, and alveolar macrophages.26 It has also been reported that cigarette smoking-induced oxidative stress is associated with apoptosis of lung epithelial cells in vitro.27 Oxidative damage from cigarette smoke is likely the initial insult, which is then followed by inflammation, apoptosis, and enlarged air space indicating emphysematous change.28 To date, many studies have reported that mainstream cigarette smoking induces apoptosis of bronchiolar and alveolar epithelial cells. But there are no reports about the relationship between the sidestream smoke and apoptosis. Our results established that sidestream smoke induces apoptosis in bronchiolar and alveolar epithelial cells on the LM and EM TUNEL staining.
- Cleavage of PARP protein is a marker that is commonly found to detect the apoptosis. In addition, it has also been reported that levels of cleaved PARP are significantly higher in smokers than in ex-smokers or non-smoking glaucomatous groups of the same gender.29 Our results showed that the increased levels of PARP cleavages were associated with the increased volume of an exposure to sidestream smoke in groups 1, 2, and 3. In groups 4 and 5 where the experimental rats were subjected to a long-term sidestream smoke, the level of PARP cleavage were increased compared to the control group on the western blot analysis.
- In conclusion, our results indicate that sidestream smoke causes bronchiolar and alveolar epithelial cell injury and this leads to alterations in bronchiolar mucosal cells as well as the early bronchiolocentric emphysematous change. Moreover, it was also shown that the severity of bronchiolar and alveolar damage was correlated with the volume and the duration of exposure to sidestream smoke.
DISCUSSION
Acknowledgments
Acknowledgments
- 1. Bach PB. Smoking as a factor in causing lung cancer. JAMA 2009; 301: 539-541. ArticlePubMed
- 2. Pelkonen M. Smoking: relationship to chronic bronchitis, chronic obstructive pulmonary disease and mortality. Curr Opin Pulm Med 2008; 14: 105-109. ArticlePubMed
- 3. Caminati A, Harari S. Smoking-related interstitial pneumonias and pulmonary Langerhans cell histiocytosis. Proc Am Thorac Soc 2006; 3: 299-306. ArticlePubMed
- 4. Beasley MB. Smoking-related Small airway disease: a review and update. Adv Anat Pathol 2010; 17: 270-276. ArticlePubMed
- 5. Vassallo R, Ryu JH. Tobacco smoke-related diffuse lung diseases. Semin Respir Crit Care Med 2008; 29: 643-650. ArticlePubMed
- 6. He Y, Lam TH, Jiang B, et al. Passive smoking and risk of peripheral arterial disease and ischemic stroke in Chinese women who never smoked. Circulation 2008; 118: 1535-1540. ArticlePubMed
- 7. Smith DR, Beh EJ. Hirayama, passive smoking and lung cancer: 30 years on and the numbers still don't lie. Public Health 2011; 125: 179-181. ArticlePubMed
- 8. Jiménez-González C, Santini V, Figueroa Cosme WI, Parilla Idel C. Do parents know about the adverse effects of passive smoking and the relationship with respiratory illness on their children? Bol Asoc Med P R 2008; 100: 39-46.
- 9. Brownson RC, Figgs LW, Caisley LE. Epidemiology of environmental tobacco smoke exposure. Oncogene 2002; 21: 7341-7348. ArticlePubMedPDF
- 10. Wagener TL, Gregor KL, Busch AM, McQuaid EL, Borrelli B. Risk perception in smokers with children with asthma. J Consult Clin Psychol 2010; 78: 980-985. ArticlePubMedPMC
- 11. Pang B, Wang C, Weng X, et al. Lung injury caused by passive smoking and its effects on cytokines in rats. Zhonghua Yu Fang Yi Xue Za Zhi 2000; 34: 104-105. PubMed
- 12. Cendon SP, Battlehner C, Lorenzi Filho G, et al. Pulmonary emphysema induced by passive smoking: an experimental study in rats. Braz J Med Biol Res 1997; 30: 1241-1247. ArticlePubMed
- 13. Kwon KY, Kim YH, Yi BY, Kang YN, Lee SS. Ultrastructural examination of early changes of small airway and pulmonary parenchyma by side-stream cigarette smoking. Keimyung Med J 2001; 20: 188-198.
- 14. Tuder RM, Petrache I, Elias JA, Voelkel NF, Henson PM. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol 2003; 28: 551-554. ArticlePubMed
- 15. Zhang C, Cai S, Chen P, et al. Inhibition of tumor necrosis factor-alpha reduces alveolar septal cell apoptosis in passive smoking rats. Chin Med J (Engl) 2008; 121: 597-601. PubMed
- 16. Marwick JA, Kirkham P, Gilmour PS, Donaldson K, MacNEE W, Rahman I. Cigarette smoke-induced oxidative stress and TGF-beta1 increase p21waf1/cip1 expression in alveolar epithelial cells. Ann N Y Acad Sci 2002; 973: 278-283. PubMed
- 17. Janerich DT, Thompson WD, Varela LR, et al. Lung cancer and exposure to tobacco smoke in the household. N Engl J Med 1990; 323: 632-636. ArticlePubMed
- 18. Nagai A, Thurlbeck WM. Scanning electron microscopic observations of emphysema in humans: a descriptive study. Am Rev Respir Dis 1991; 144: 901-908. ArticlePubMed
- 19. Watanabe N, Fukushima M, Taniguchi A, et al. Smoking, white blood cell counts, and TNF system activity in Japanese male subjects with normal glucose tolerance. Tob Induc Dis 2011; 9: 12.ArticlePubMedPMC
- 20. Zoppini G, Faccini G, Muggeo M, Zenari L, Falezza G, Targher G. Elevated plasma levels of soluble receptors of TNF-alpha and their association with smoking and microvascular complications in young adults with type 1 diabetes. J Clin Endocrinol Metab 2001; 86: 3805-3808. PubMed
- 21. Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl 2001; 34: 50s-59s. Article
- 22. Corroyer S, Maitre B, Cazals V, Clement A. Altered regulation of G1 cyclins in oxidant-induced growth arrest of lung alveolar epithelial cells: accumulation of inactive cyclin E-DCK2 complexes. J Biol Chem 1996; 271: 25117-25125. PubMed
- 23. Singh A, Kapur S, Chattopadhyay I, et al. Cytokeratin immunoexpression in esophageal squamous cell carcinoma of high-risk population in Northeast India. Appl Immunohistochem Mol Morphol 2009; 17: 419-424. ArticlePubMedPMC
- 24. Xue LY, Hu N, Song YM, et al. Tissue microarray analysis reveals a tight correlation between protein expression pattern and progression of esophageal squamous cell carcinoma. BMC Cancer 2006; 6: 296.ArticlePubMedPMCPDF
- 25. Lee JJ, Liu D, Lee JS, et al. Long-term impact of smoking on lung epithelial proliferation in current and former smokers. J Natl Cancer Inst 2001; 93: 1081-1088. ArticlePubMed
- 26. Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2001; 281: L1392-L1401. ArticlePubMed
- 27. Ramage L, Jones AC, Whelan CJ. Induction of apoptosis with tobacco smoke and related products in A549 lung epithelial cells in vitro. J Inflamm (Lond) 2006; 3: 3.ArticlePubMedPMCPDF
- 28. Banerjee S, Maity P, Mukherjee S, et al. Black tea prevents cigarette smoke-induced apoptosis and lung damage. J Inflamm (Lond) 2007; 4: 3.ArticlePubMedPMCPDF
- 29. Zanon-Moreno V, Garcia-Medina JJ, Zanon-Viguer V, Moreno-Nadal MA, Pinazo-Duran MD. Smoking, an additional risk factor in elder women with primary open-angle glaucoma. Mol Vis 2009; 15: 2953-2959. PubMedPMC
REFERENCES
Exposure to side-stream smoke: group 1, 3 cigarettes/day for 1 mo; group 2, 5 cigarettes/day for 1 mo; group 3, 7 cigarettes/day for 1 mo; group 4, 5 cigarettes/day for 3 mo; group 5, 5 cigarettes/day for 6 mo.
TNF-α, tumor necrosis factor α; TGF-β1, tumor growth factor β1; IL, interleukin; CK, cytokeratin; A, alveolar cells; B, bronchiolar cells.
aNuclear expression: scoring 0, positive expression ≤5%; 1, 6-20%; 2, 21-40%; 3, 41-70%; 4, >70%.
Figure & Data
References
Citations
- Cigarette smoke extract alters the cell cycle via the phospholipid transfer protein/transforming growth factor-β1/CyclinD1/CDK4 pathway
Xue-Min Chai, You-Lun Li, Hong Chen, Shu-Liang Guo, Li-Li Shui, Ya-Juan Chen
European Journal of Pharmacology.2016; 786: 85. CrossRef - Keratin-14 Expression in Pneumocytes as a Marker of Lung Regeneration/Repair during Diffuse Alveolar Damage
Miriam Ficial, Caterina Antonaglia, Marco Chilosi, Mario Santagiuliana, Al-Omoush Tahseen, Davide Confalonieri, Lorenzo Zandonà, Rossana Bussani, Marco Confalonieri
American Journal of Respiratory and Critical Care Medicine.2014; 189(9): 1142. CrossRef
 PubReader
PubReader Cite this Article
Cite this Article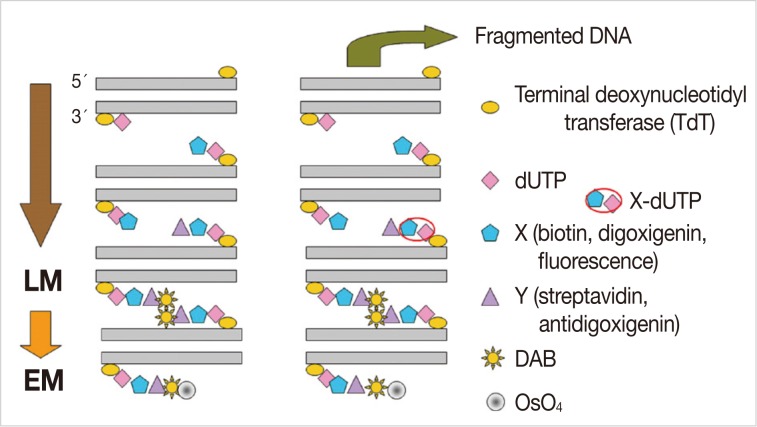
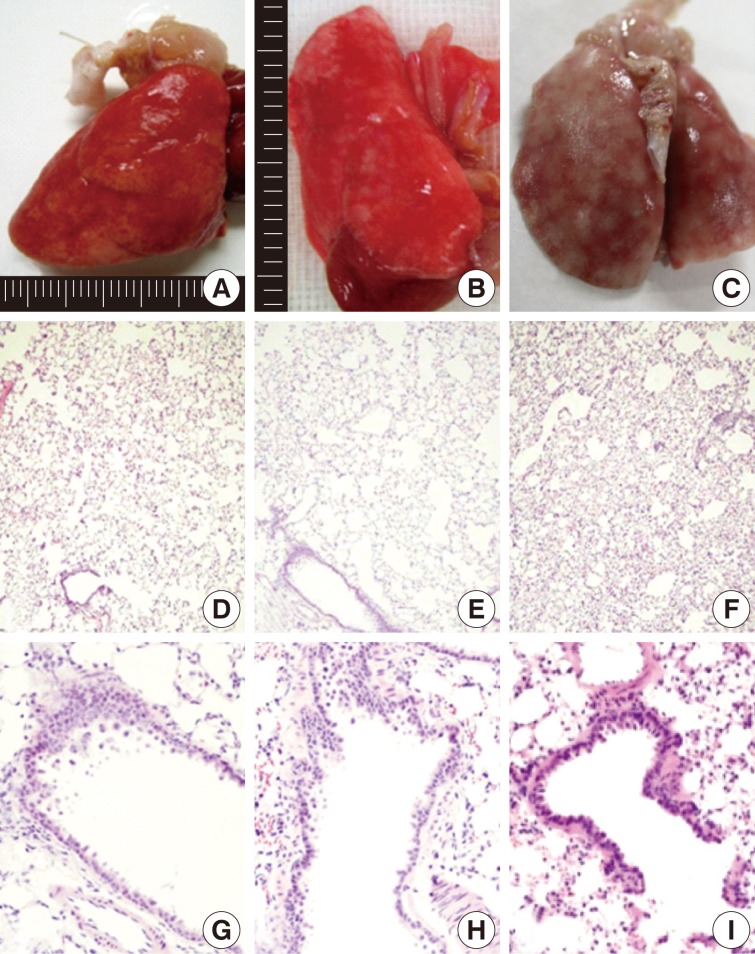
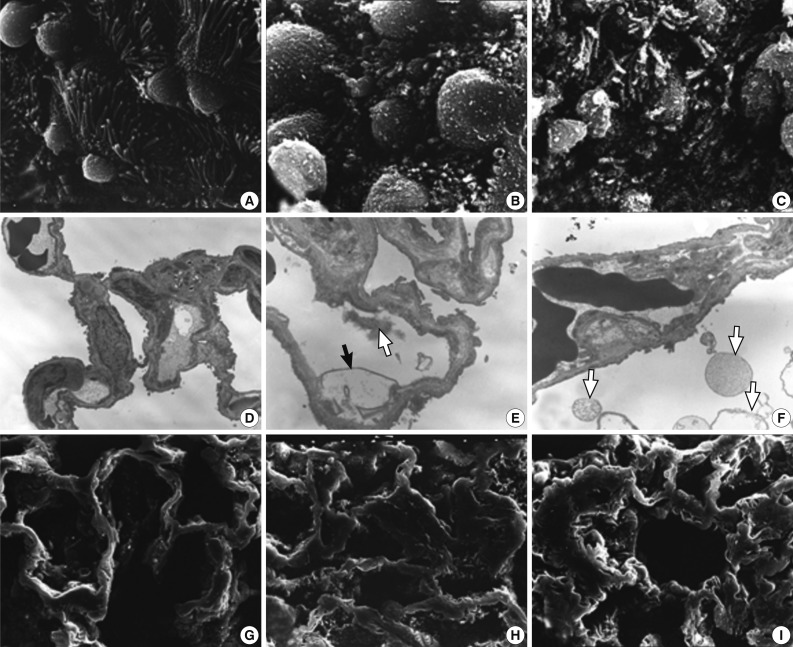
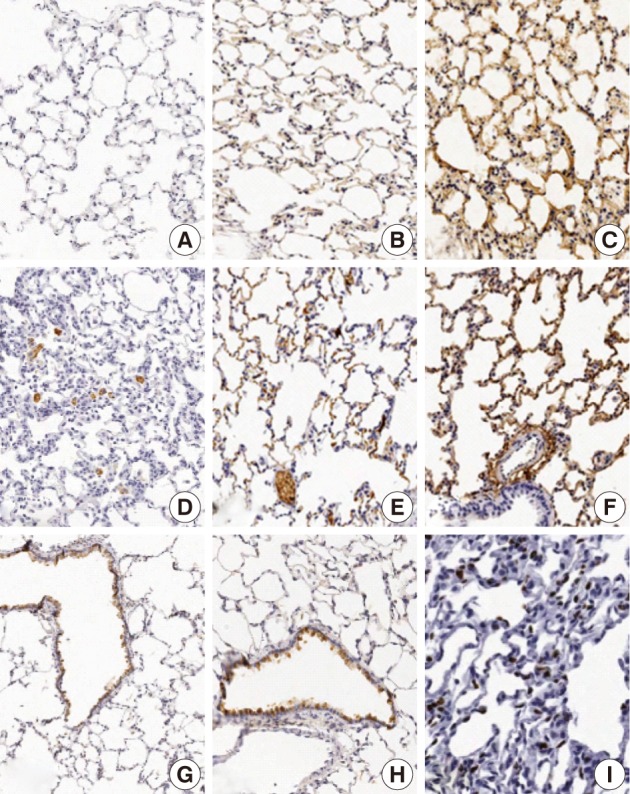
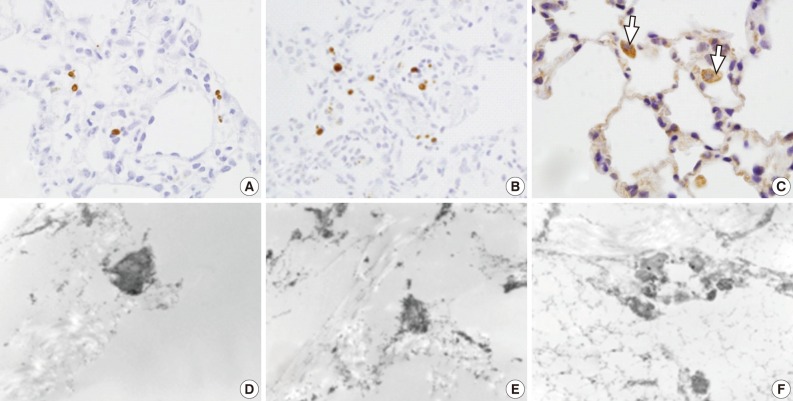







Fig. 1
Fig. 2
Fig. 3
Fig. 4
Fig. 5
Fig. 6
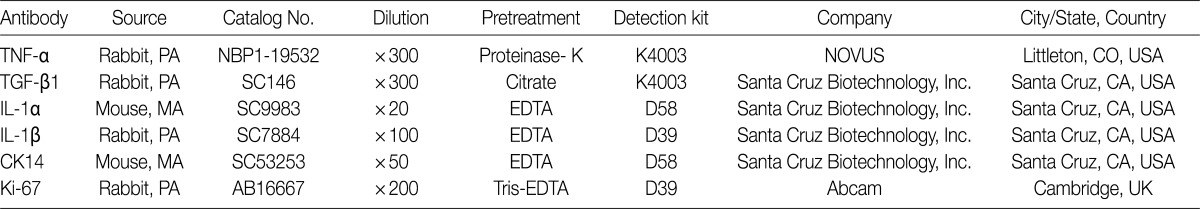
TNF-α, tumor necrosis factor α; PA, polyclonal antibody; TGF-β1, tumor growth factor β1; MA, monoclonal antibody; IL, interleukin; EDTA, ethylenediaminetetraacetic acid; CK, cytokeratin.
Exposure to side-stream smoke: group 1, 3 cigarettes/day for 1 mo; group 2, 5 cigarettes/day for 1 mo; group 3, 7 cigarettes/day for 1 mo; +/-, minimal; +, mild; ++, moderate; +++, prominent.
Exposure to side-stream smoke: group 2, 5 cigarettes/day for 1 mo; group 4, 5 cigarettes/day for 3 mo; group 5, 5 cigarettes/day for 6 mo; +/-, minimal; +, mild; ++, moderate; +++, prominent.
Exposure to side-stream smoke: group 1, 3 cigarettes/day for 1 mo; group 2, 5 cigarettes/day for 1 mo; group 3, 7 cigarettes/day for 1 mo; group 4, 5 cigarettes/day for 3 mo; group 5, 5 cigarettes/day for 6 mo. TNF-α, tumor necrosis factor α; TGF-β1, tumor growth factor β1; IL, interleukin; CK, cytokeratin; A, alveolar cells; B, bronchiolar cells. aNuclear expression: scoring 0, positive expression ≤5%; 1, 6-20%; 2, 21-40%; 3, 41-70%; 4, >70%.

