 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 60(2); 2026 > Article
-
Review Article
Cutaneous soft tissue tumors in the 5th edition of the World Health Organization classification of skin tumors: key updates and new entities -
Joon Hyuk Choi

-
Journal of Pathology and Translational Medicine 2026;60(2):144-183.
DOI: https://doi.org/10.4132/jptm.2026.01.09
Published online: March 13, 2026
Department of Pathology, Yeungnam University College of Medicine, Daegu, Korea
- Corresponding Author: Joon Hyuk Choi, MD, PhD Department of Pathology, Yeungnam University College of Medicine, 170 Hyeonchung-ro, Namgu, Daegu 42415, Korea Tel: +82-53-640-6754, Fax: +82-53-640-6769, E-mail: joonhyukchoi@ynu.ac.kr
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 3,370 Views
- 238 Download
Abstract
- The 5th edition of the World Health Organization (WHO) classification of skin tumors introduces a dedicated chapter on cutaneous soft tissue tumors, providing a comprehensive, standardized reference with updated diagnostic criteria that directly inform routine dermatopathology practice and molecular diagnostics. This edition incorporates several key changes, including newly recognized entities such as EWSR1::SMAD3-rearranged fibroblastic tumor, neurotrophic tyrosine receptor kinase (NTRK)–rearranged spindle cell neoplasm, superficial CD34-positive fibroblastic tumor, and CRTC1::TRIM11 cutaneous tumor. Diagnostic terminology has also been refined; for example, the term ‘atypical intradermal smooth muscle neoplasm’ replaces ‘cutaneous leiomyosarcoma’ for lesions confined to the dermis, whereas the designation leiomyosarcoma is reserved for tumors with overt subcutaneous infiltration. In addition, epithelioid fibrous histiocytoma has been reassigned to the family of tumors of uncertain differentiation. This review summarizes the key updates and newly recognized entities in the chapter on cutaneous soft tissue tumors in the 5th edition of the WHO classification of skin tumors, emphasizing their clinicopathological and molecular implications.
- The 5th edition of the World Health Organization (WHO) classification of skin tumors, volume 12 of the WHO classification of tumors series, was published online in 2023 and in print in September 2025 [1]. This series is widely regarded as the gold standard for tumor diagnosis, providing an authoritative synthesis of clinical, histopathological, and molecular criteria, as well as practical diagnostic approaches. Since the publication of the 4th edition of the WHO classification of skin tumors in 2018 [2], significant advances have been made in soft tissue pathology, particularly in identifying novel molecular and genetic alterations, discovering new immunohistochemical markers, and clarifying underlying biological mechanisms [3,4]. The 5th edition incorporates these advances, thereby reflecting major progress in the understanding of soft tissue tumors.
- The dedicated chapter on soft tissue tumors in the 5th edition of the WHO classification of skin tumors includes entities that are confined to the skin or characterized by distinctive cutaneous features. This chapter maintains the organizational structure of the previous edition, classifying soft tissue tumors into seven main families: (1) adipocytic tumors, (2) fibroblastic, myofibroblastic, and fibrohistiocytic tumors, (3) vascular tumors, (4) pericytic and perivascular tumors, (5) smooth muscle tumors, (6) neural tumors, and (7) tumors of uncertain differentiation (Table 1).
- Cutaneous soft tissue tumors present considerable diagnostic challenges due to their diversity and overlapping histological features. The 5th edition recognizes several newly defined entities that reflect recent advances in tumor biology, molecular genetics, and diagnostic approaches [5,6]. A thorough understanding of these updates is essential for accurate diagnosis, optimal patient management, and improved prognostication.
- This review summarizes key updates to cutaneous soft tissue tumors in the 5th edition of the WHO classification, highlighting newly recognized entities and tumors incorporated from other WHO volumes, along with their clinical, histological, and molecular features, and recent advances in tumor pathogenesis and immunohistochemistry (IHC). It is intended to serve as a practical reference for practicing pathologists, with particular emphasis on updated diagnostic criteria and newly introduced molecular correlates.
- The updates in the 5th edition of the WHO classification of skin tumors are summarized as follows: newly recognized tumor entities and tumors incorporated from other WHO volumes (Table 2); newly identified molecular genetic alterations and immunohistochemical markers (Table 3); tumors with revised terminology (Table 4); and tumors reassigned to different families (Table 5).
INTRODUCTION
- Key updates
- In the 5th WHO classification, nevus lipomatosus superficialis is defined as a benign cutaneous hamartoma characterized by dermal ectopic adipose tissue rather than as a developmental anomaly [7]. It is classified into two subtypes: classic and solitary. Additionally, among conventional lipomas, osteolipoma and chondrolipoma are recognized as distinct subtypes, defined by osseous and cartilaginous metaplasia, respectively [8]. In contrast, previously described subtypes, including fibrolipoma, myxolipoma, angiomyxolipoma, sclerotic lipoma, and chondroid lipoma, have been removed from the current WHO classification [9]. Cellular angiolipoma is defined as a subtype with a predominant vascular component and a sparse adipocytic component [10,11]. Spindle cell lipoma (SCL) and pleomorphic lipoma (PL) represent a morphological spectrum of the same benign adipocytic neoplasm [12], sharing RB1 deletion at chromosome 13q14, supporting their close biological relationship. Recognized morphologic patterns include fat-poor, myxoid, and pseudoangiomatous variants of SCL/PL (Fig. 1A, B).
- Atypical lipomatous tumor (ALT) is defined as an adipocytic neoplasm of borderline malignancy characterized by focal nuclear atypia and MDM2 and/or CDK4 amplification [13]. Primary cutaneous or subcutaneous ALTs are exceedingly rare [14,15]. Mature heterologous elements, such as osseous or myogenic tissue, may occasionally be present; this does not indicate dedifferentiation [16,17]. The terms ALT and well-differentiated liposarcoma (WDLPS) are synonymous, as they describe morphologically and genetically identical neoplasms. However, the term ‘WDLPS’ should be avoided for superficial tumors because these lesions typically exhibit less aggressive behavior. Pleomorphic liposarcoma (PLPS) is a high-grade, pleomorphic spindle cell sarcoma containing variable numbers of pleomorphic lipoblasts without components of WDLPS or other differentiation (Fig. 2A, B) [18]. An epithelioid subtype of PLPS, characterized by epithelioid cytomorphology, has been recognized as a distinct subtype [19]. The absence of MDM2 amplification is a key diagnostic criterion distinguishing PLPS from ALT and dedifferentiated liposarcoma.
ADIPOCYTIC TUMORS
- Key updates
- The fibroblastic, myofibroblastic, and fibrohistiocytic tumor family has been expanded in the 5th WHO classification to include several newly described entities, such as fibrous papule, fibroblastic connective tissue nevus (FCTN), fibro-osseous tumor of digits (FOTD), inclusion body fibromatosis (IBF), multinucleate cell angiohistiocytoma (MCA), and EWSR1::SMAD3-rearranged fibroblastic tumor (ESRFT). Epithelioid fibrous histiocytoma (EFH), previously classified as a subtype of dermatofibroma (fibrous histiocytoma), is now recognized as a distinct entity characterized by anaplastic lymphoma kinase (ALK) overexpression due to ALK rearrangement and reassigned to the family of tumors of uncertain differentiation. Myxofibrosarcoma, previously placed in the family of tumors of uncertain differentiation, has been reclassified within the fibroblastic, myofibroblastic, and fibrohistiocytic tumor family. The fibroblastic tumor previously designated as EWSR1::SMAD3-positive (emerging) is now formally recognized as an ESRFT.
- Additionally, the 5th WHO classification identifies recurrent USP6 gene rearrangements with multiple fusion partners, including PKM, RCC1, ASPN, COL1A1, COL3A1, MYH9, MIR22HG, CTNNB1, SPARC, CAP1, EMP1, CYTOR, NR1D1, RAB1A, and TNC, in fibroma of tendon sheath [20,21], supporting the concept that some cases represent a tenosynovial variant of nodular fasciitis [22]. Calcifying aponeurotic fibroma (CAF) harbors a recurrent FN1::EGF fusion, leading to aberrant epidermal growth factor (EGF) expression [23]. This fusion has also been identified in a subset of lipofibromatosis-like tumors, which may represent CAF variants lacking calcification [24]. Patients younger than 5 years are at higher risk of local recurrence [25]. In sclerotic fibroma, loss of phosphatase and tensin homolog (PTEN) expression on IHC may suggest Cowden syndrome [26]. Multiple pleomorphic fibromas in young patients may indicate an underlying genetic cancer syndrome, such as Li-Fraumeni syndrome associated with TP53 germline mutations [27].
- In the 5th edition, dermatofibroma (fibrous histiocytoma) is formally classified into five subtypes—classic, cellular, aneurysmal, atypical, and deep fibrous histiocytoma—each with distinct clinical and pathological features (Fig. 3A, B) [28]. Recurrent gene fusions involving PKC isoforms (PRKCA, PRKCB, and PRKCD) and various genes encoding membrane-associated proteins (LAMTOR1, PDPN, CD63, and KIRREL family genes) have been identified in a subset of these tumors [29-31]. Superficial fibromatosis is defined as a nodular, locally recurrent fibroblastic/myofibroblastic proliferation that includes palmar, plantar, and penile subtypes [32]. Although superficial fibromatosis involves activation of the WNT/β-catenin signaling pathway, it lacks the CTNNB1 and APC mutations characteristic of desmoid fibromatosis [33]. Plexiform fibrohistiocytic tumor exhibits diverse cytogenetic alterations, including 46,X,del(X)(q13)[3]/46,XX[23]; 46,XY,t(4;15)(q21;q15); and 46,XY,-6,-8,del(4)(q25q31),del(20)(q11.2) with additional der(8)(p22) [34-36].
- In cutaneous myxoma (superficial angiomyxoma), approximately 45%–65% of Carney complex-associated cases harbor PRKAR1A mutations [37,38], while alternative genetic pathways, including MYH8 mutations, have also been identified [39]. Nodular fasciitis shows USP6 rearrangements in approximately 90% of cases, with MYH9::USP6 accounting for >65% of these [40]. Atypical or malignant nodular fasciitis cases harboring PPP6R3::USP6 or CALD1::USP6 fusions have rarely been reported [41,42]. An EIF5A::USP6 fusion has also been identified in dermal nodular fasciitis of the forearm with atypical mitoses [43]. In dermatofibrosarcoma protuberans (DFSP), COL1A1::PDGFB remains the hallmark translocation, while alternative PDGFD rearrangements involving COL6A3, EMILIN2, or TNC have also been described [44-46]. Genomic gains of COL1A1::PDGFB and alterations in the mechanistic target of rapamycin (mTOR) pathway have been associated with fibrosarcomatous transformation [46,47]. The subtypes of DFSP have been standardized to include classic DFSP, giant cell fibroblastoma, myxoid DFSP, pigmented DFSP, myoid DFSP, plaque-like DFSP, DFSP with fibrosarcomatous transformation, and other morphological patterns (granular cell, sclerosing, and atrophic types) (Fig. 4A, B) [48].
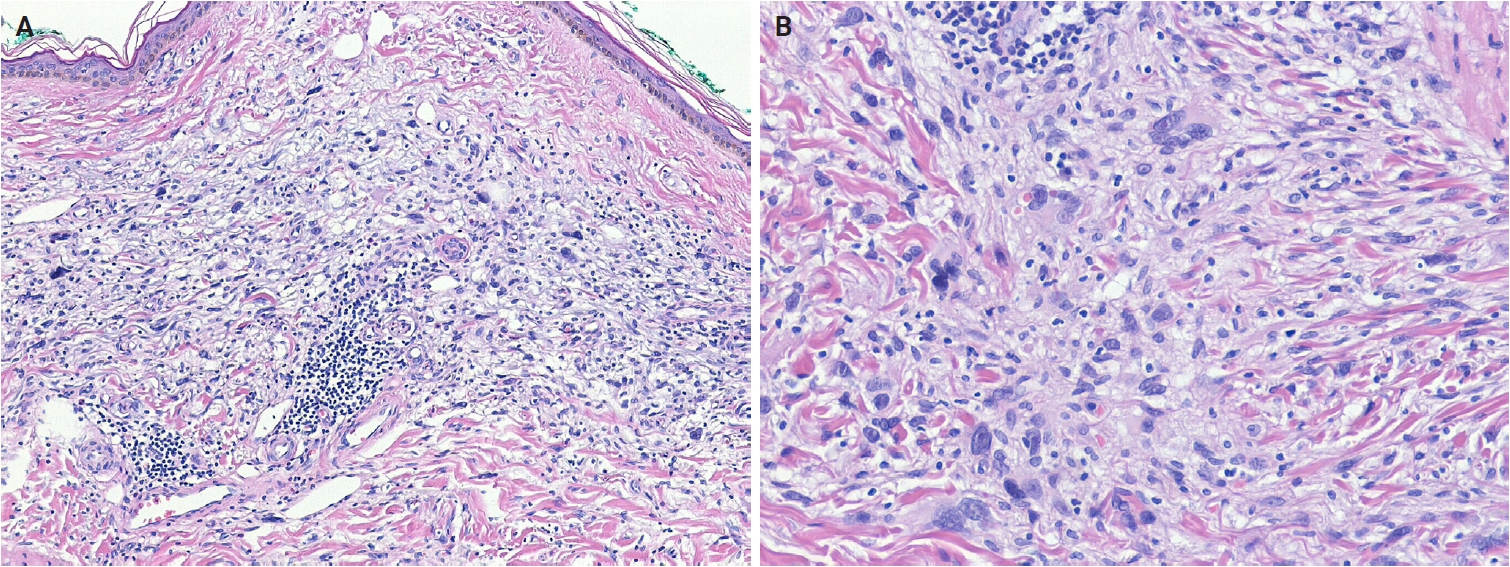
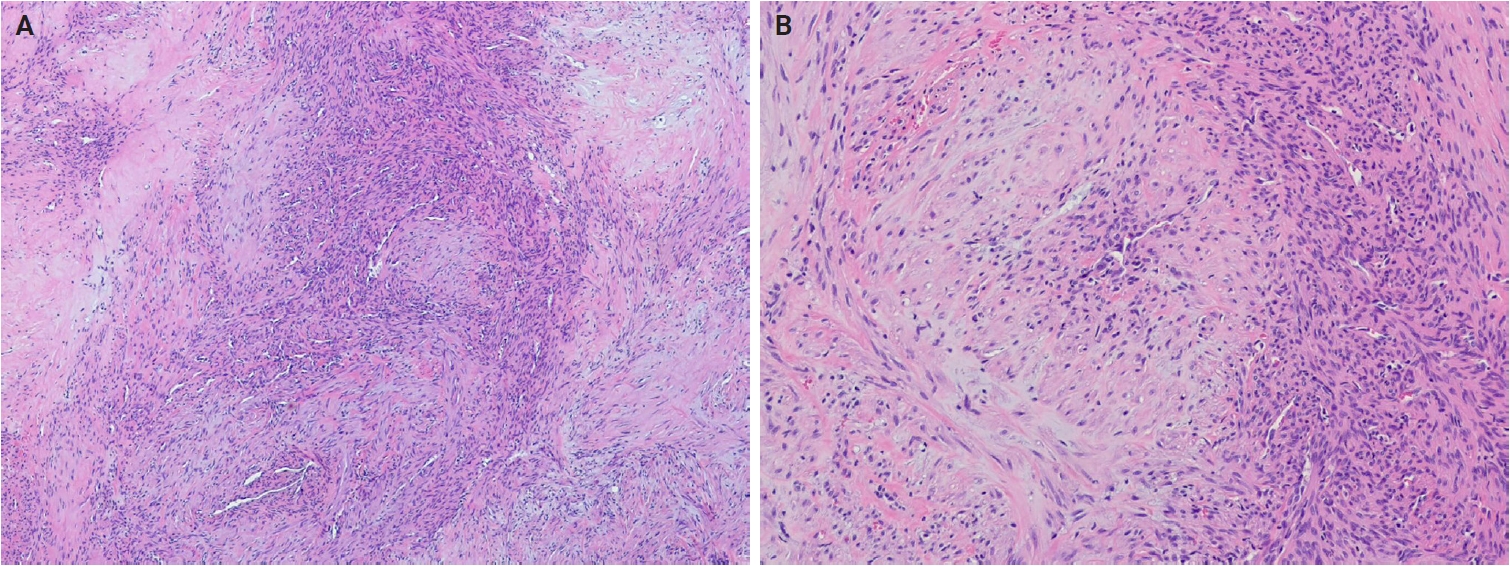
- In myxoinflammatory fibroblastic sarcoma (MIFS), VGLL3 amplification at 3p11-p12 occurs in >50% of cases, resulting in a supernumerary ring chromosome [49]. Histologically, MIFS is characterized by atypical fibroblastic cells with macronucleoli and a mixed inflammatory infiltrate in variably myxoid and hyalinized stroma (Fig. 5A, B) [50]. A t(1;10) translocation involving MGEA5 (OAG) (10q24) and TGFBR3 (1p22) is present in approximately 13% of cases and has also been identified in pure hemosiderotic fibrolipomatous tumors (HFLTs), hybrid MIFS-HFLT lesions, and pleomorphic hyalinizing angiectatic tumors [51-53]. BRAF fusions or amplifications have also been identified [54]. Myxofibrosarcomas exhibit frequent chromosomal rearrangements and copy-number alterations, even in low-grade tumors, with increasing genomic complexity in recurrent tumors showing histological progression [55-58]. Myxofibrosarcomas are characterized by somatic copy-number alterations, with several oncogenes—TRIO, RICTOR, SKP2, and AMACR—located on 5p, which is frequently gained, often co-amplified, and associated with higher histological grade [59-61]. Alterations in tumor suppressor genes, including TP53 (46%), RB1 (18%), and CDKN2A/CDKN2B (16%), occur more frequently than oncogene amplifications involving CDK6, CCND1, or MDM2 [61].
- Fibrous papule
- Fibrous papule is a benign lesion of presumed fibroblastic origin that most commonly arises on the nose or central face [62]. It predominantly affects middle-aged adults [63]. Clinically, it presents as a solitary, sporadic, dome-shaped, sessile papule. The presence of multiple (syndromic) fibrous papules is associated with tuberous sclerosis and, less commonly, with Birt-Hogg-Dubé syndrome or multiple endocrine neoplasia type 1 [64,65]. In tuberous sclerosis, multiple fibrous papules are associated with germline mutations in TSC1 or TSC2, leading to activation of the mTOR pathway. A similar mTOR-related mechanism has also been proposed for sporadic fibrous papules [66].
- Fibrous papules are well-circumscribed papular lesions with a tan to white cut surface. Histologically, they are dome-shaped and composed of dilated dermal blood vessels, surrounded by fibrotic or collagenized stroma containing spindle-shaped or stellate fibroblasts [67]. The overlying epidermis may exhibit melanocytic hyperplasia [68]. Histological subtypes include hypercellular, pigmented, pleomorphic, clear cell, granular cell, epithelioid, and inflammatory variants.
- Fibroblastic connective tissue nevus
- FCTN is a benign, dermal mesenchymal lesion of the fibroblastic/myofibroblastic lineage, characterized by short, intersecting fascicles of bland spindle cells [69]. Lesions commonly occur on the trunk, head and neck, or extremities [70-72]. FCTN shows a slight female predominance and typically affects children and young individuals (median age, 10 years). Congenital cases have been reported, often presenting as large, infiltrated plaques on the trunk [73,74]. Clinically, FCTN usually manifests as a solitary, slowly growing, painless plaque or nodule, although rare presentations include agminate lesions [75,76], atrophic plaques [77], or exophytic tumors. A single case harboring the KHDRBS1::NTRK3 gene fusion has been reported [73].
- Histologically, FCTN is a poorly circumscribed, dermal-based lesion localized primarily in the reticular dermis and often extending into the superficial subcutis. It comprises bland, spindle-shaped to ovoid cells arranged in short, intersecting fascicles, with preserved adnexal structures. Cellularity varies, mitotic figures are rare or absent, and a storiform pattern may occasionally be observed. Epidermal papillomatosis and ectopic dermal adipose tissue are frequently present [70]. Elastic fibers often decrease. Immunohistochemically, tumor cells express CD34 and smooth muscle actin (SMA), supporting fibroblastic/myofibroblastic differentiation [71].
- Fibro-osseous tumor of digits
- FOTD is a localized, self-limiting benign neoplasm that arises in the soft tissue of the digits [78]. It most commonly occurs around the proximal phalanx of the fingers and, less frequently, the toes [79-81]. FOTD occurs over a wide age range, with a predilection for young adults [79,80]. Clinically, it presents as a rapidly growing mass that may be painful and demonstrates irregular calcifications on imaging. Over time, the lesion hardens and becomes well-circumscribed [82]. Molecular studies have identified USP6 fusion genes in FOTD [80,83], a feature shared with nodular fasciitis, which shows overlapping morphology.
- Histologically, FOTD shows nodular fasciitis-like areas with variable stages of bone formation. The lesion is hypercellular and composed of randomly arranged or short, intersecting fascicles of spindle cells with plump, fusiform nuclei containing fine chromatin, conspicuous nucleoli, and moderate amounts of eosinophilic cytoplasm. Mitoses are frequent but typical. The stroma is richly vascular and variably myxoid, containing fibrin, extravasated erythrocytes, scattered lymphocytes, and osteoclast-like giant cells. Spindle cells gradually blend into ill-defined trabeculae and sheets of unmineralized woven bone, which are rimmed by prominent osteoblasts with amphophilic cytoplasm and contain large osteocytes [82,84].
- Inclusion body fibromatosis
- IBF is a benign myofibroblastic tumor characterized by eosinophilic intracytoplasmic inclusions and a propensity for local recurrence [85]. It is subclassified into classic (digital) and non-classic (extradigital) subtypes. Classic IBF accounts for approximately 2% of pediatric fibroblastic tumors [86], with >80% of cases diagnosed before the age of three and approximately 30% present at birth [87]. There is no sex predilection. Rare adult cases have been reported [88]. Classic IBFs typically arise on the dorsal or dorsolateral aspects of the distal or middle phalanges of the second to fourth digits, less frequently the fifth digit, and usually spare the first digit [89]. Non-classic IBF involves the extremities, tongue, and breast [90]. Clinically, classic IBFs present as asymptomatic, dome-shaped or polypoid nodules, typically <20 mm in diameter [89]. They may occur synchronously or metachronously, occasionally involving multiple digits [91]; concurrent finger and toe involvement is rare. In contrast, non-classic IBFs usually present as a solitary mass or nodule.
- Histologically, IBF comprises spindle cells with lightly eosinophilic cytoplasm and elongated, bland nuclei, exhibiting low mitotic activity. These cells form whorls, short interlacing fascicles, or storiform patterns within the collagenous dermis, often oriented perpendicularly to the epidermis and encircling adnexal structures, with occasional extension into deeper tissues [91]. Rounded, pale pink, intracytoplasmic inclusions (1.5–24 μm) are highlighted by trichrome (red), phosphotungstic acid–hematoxylin (dark purple), and Movat’s stains (pink) [89]. Immunohistochemically, tumor cells express vimentin, muscle actins (displaying peripheral ‘tram-track’ pattern), calponin, desmin, and CD99 [92]. The inclusions are positive for calponin-1 [93] and occasionally for caldesmon [89].
- Multinucleate cell angiohistiocytoma
- MCA is a benign, acquired lesion of fibrohistiocytic and vascular lineage [94]. It most commonly occurs on the dorsal surfaces of the hands, fingers, lower extremities, trunk, and back [95], with head and neck involvement being uncommon. Clinically, MCA typically presents in middle-aged adults as solitary or multiple smooth, erythematous papules or nodules. Some studies have reported a female predominance [96], whereas others have observed no significant sex difference [97]. The pathogenesis remains uncertain; proposed mechanisms include fibroblast proliferation, estrogen receptor α overexpression, and mast cell involvement [98,99].
- Histologically, MCA exhibits a characteristic triad: multinucleated stromal cells, parallel stromal fibrosis, and proliferation of small-caliber vessels. Multinucleated stromal cells are usually angulated or stellate, with elongated cytoplasmic processes, and are often sparse, detectable only in deeper sections [97]. Stromal fibrosis runs parallel to the epidermis, and the papillary and reticular dermis contain numerous thick-walled vessels with prominent endothelial cells. Immunohistochemically, multinucleated cells variably express CD68 and factor XIIIa, but are usually negative for CD163, CD34, and factor VIII [97,100]. Endothelial markers, including CD31, CD34, ETS-related gene (ERG), and factor VIII, highlight vascular proliferation.
- EWSR1::SMAD3-rearranged fibroblastic tumor
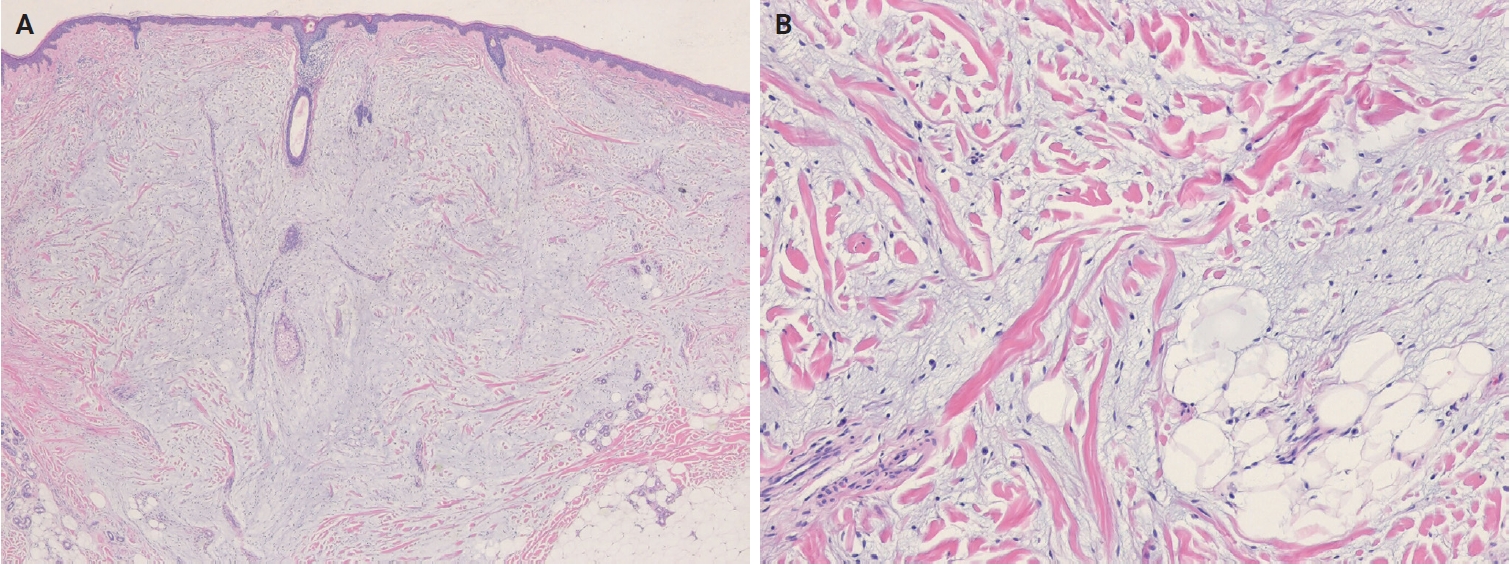
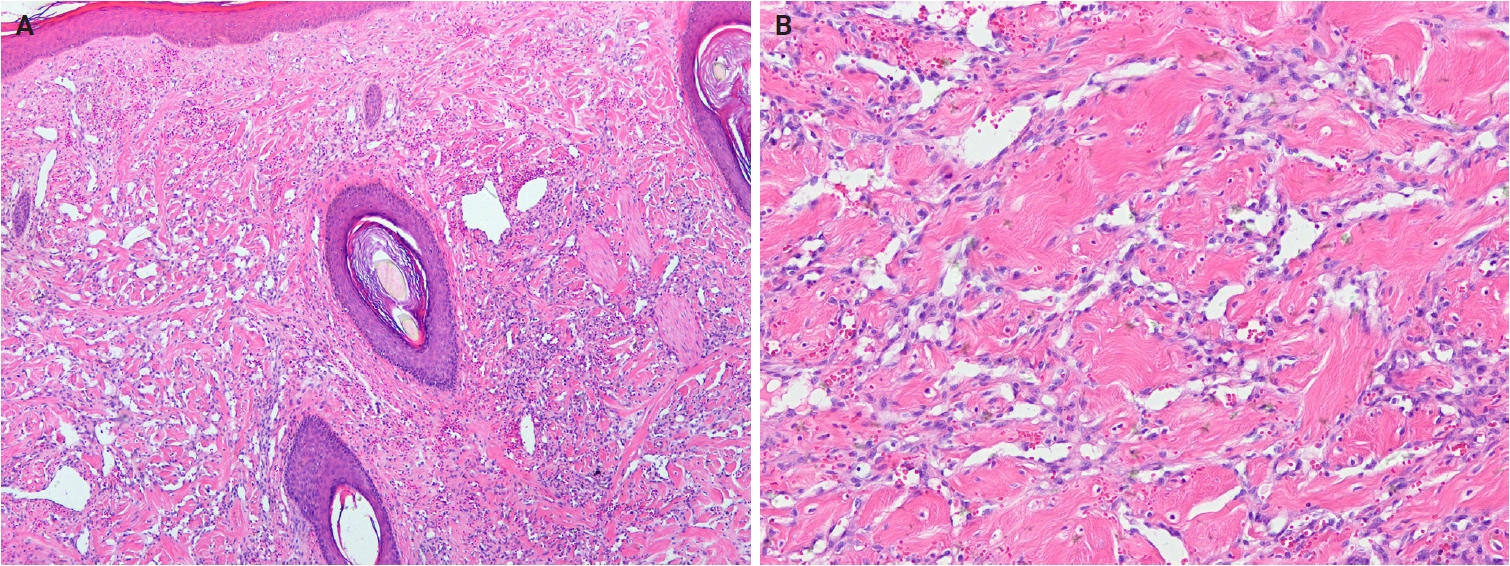
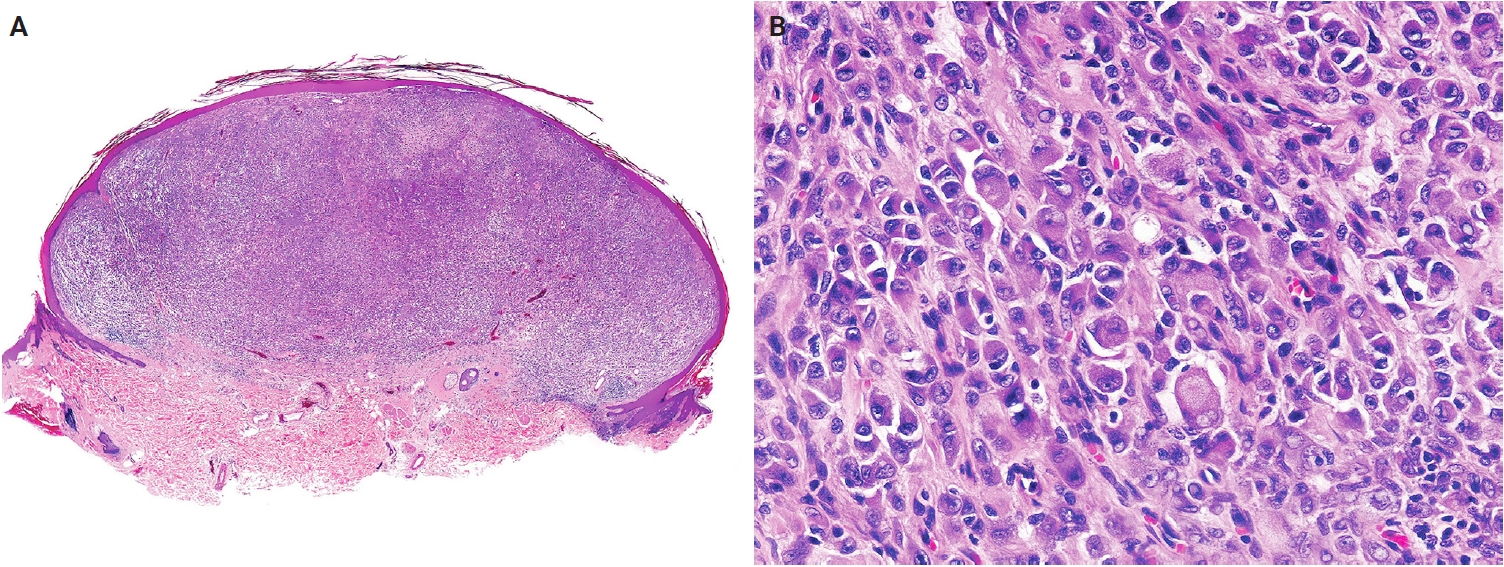
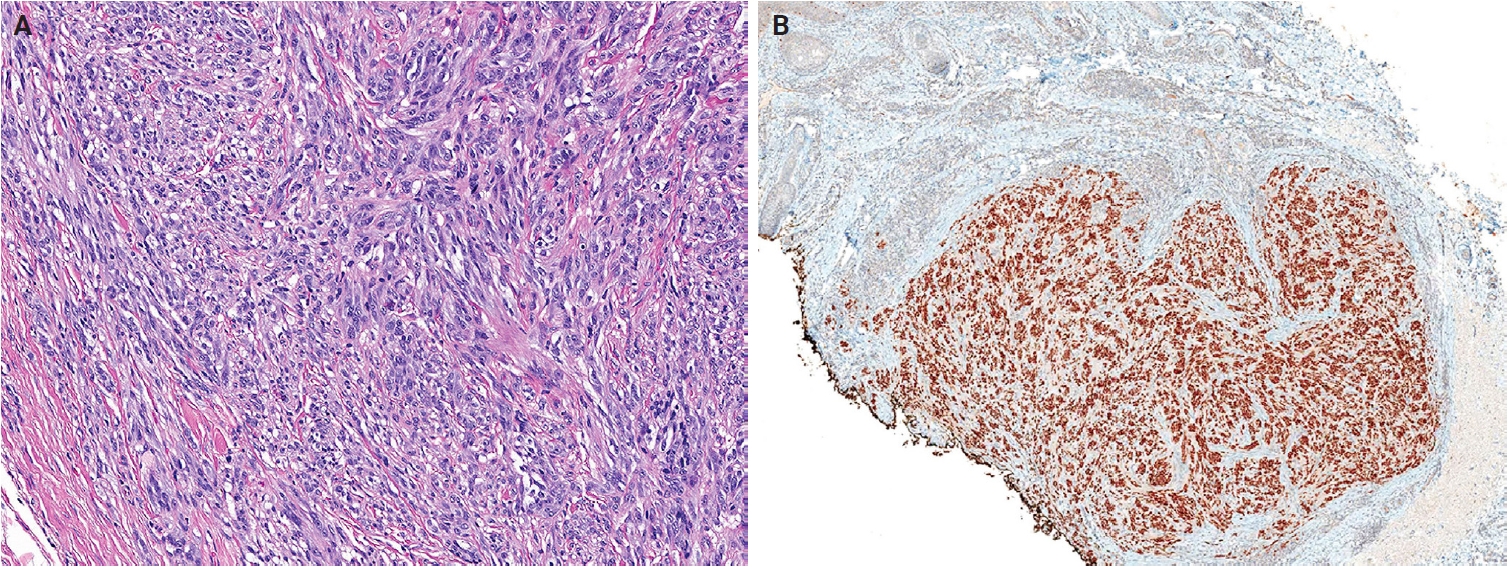
- ESRFT is a rare, distinctive benign fibroblastic neoplasm [101]. Most cases arise in the acral region, particularly the hands and feet [102], although the lower extremity is the most common non-acral site. Clinically, ESRFT presents as a small, painless, superficial nodule, typically measuring 10–20 mm in diameter. It affects a wide age range (1–68 years; median age, 39 years) and shows a female predominance (male-to-female ratio, 1:4) [103]. Molecularly, ESRFT is characterized by an EWSR1::SMAD3 gene fusion, typically involving exon 7 of EWSR1 and exon 5 or 6 of SMAD3. SMAD family member 3 (SMAD3) functions as a key signal transducer in the TGF-β/SMAD pathway and is essential for fibroblast-mediated extracellular matrix synthesis, thereby supporting the fibroblastic differentiation of this tumor.
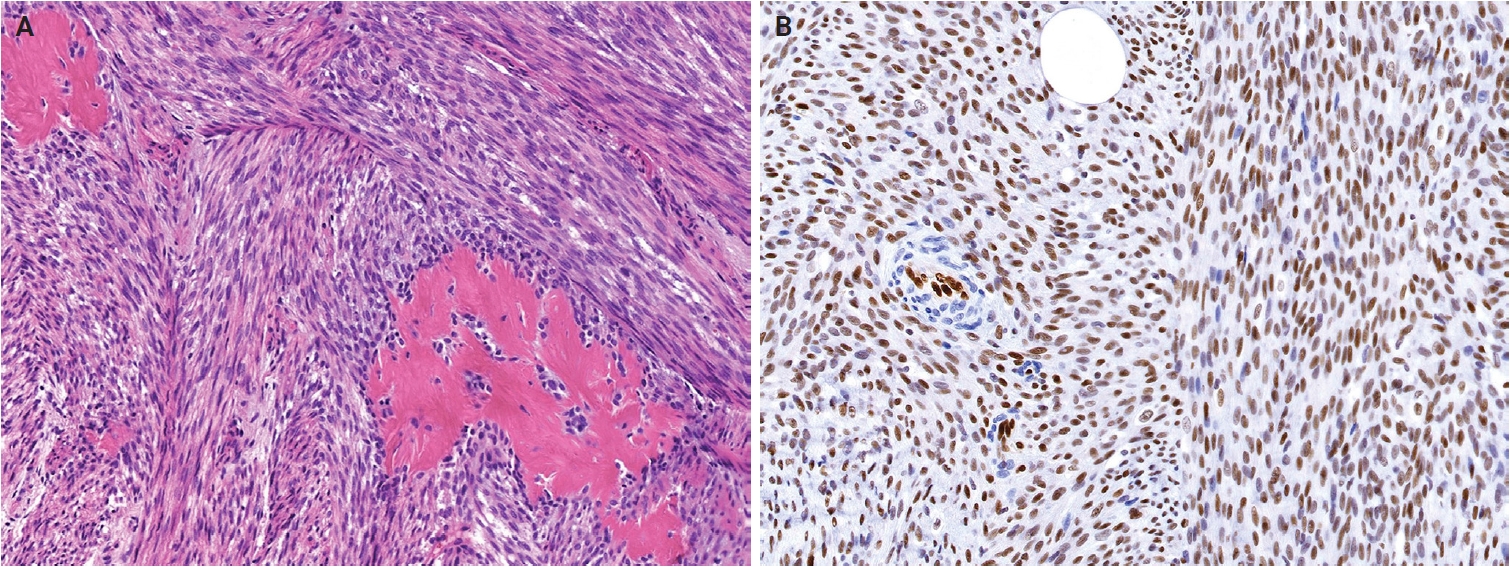
- Histologically, ESRFTs are typically well-circumscribed, although focal infiltration into the subcutaneous fat may be observed. In adults, the tumor frequently exhibits a hyalinized, acellular central zone resembling collagen rosettes. This zone is surrounded by peripheral areas composed of intersecting fascicles of spindle-shaped fibroblastic cells embedded in a collagenous to variably myxoid stroma (Fig. 6A). Approximately half of the cases lack this zonation pattern. Tumor cells are cytologically bland, with no nuclear pleomorphism, hyperchromasia, prominent nucleoli, or increased mitotic activity. Myopericytomatous growth is rare, and focal stippled dystrophic calcification may be present [102,103]. Immunohistochemically, tumor cells demonstrate strong, diffuse nuclear ERG expression, whereas SMA and CD34 are negative (Fig. 6B).
FIBROBLASTIC, MYOFIBROBLASTIC, AND FIBROHISTIOCYTIC TUMORS
- Key updates
- In the 5th WHO classification, hemangiomas and other benign vascular tumors are grouped separately. Newly described entities in the vascular tumor family include papillary hemangioma, poikilodermatous plaque-like hemangioma (PPLH), acquired elastotic hemangioma (AEH), verrucous venous malformation, and hobnail hemangioendothelioma (HHE). Tufted hemangioma, a benign cutaneous vascular tumor sometimes associated with Kasabach-Merritt syndrome, is now generally considered part of the Kaposiform hemangioendothelioma (KHE) spectrum [104,105], and KHE has been removed from this volume. The term ‘postradiation atypical vascular lesion’ replaces the previous designation ‘atypical vascular lesion’, highlighting its association with prior radiation exposure and distinguishing it from radiation-induced angiosarcoma [106]. Additionally, papillary intralymphatic angioendothelioma (PILA) and retiform hemangioendothelioma (RHE) are now unified under the designation of HHE [107].
- In cherry hemangioma, somatic mutations in HRAS and KRAS have been identified in a small subset of cases [108]. The 5th edition also describes frequent somatic mutations in GNA14, GNA11, and GNAQ [109,110]. In glomeruloid hemangioma, elevated serum vascular endothelial growth factor levels suggest a potential role in lesion development [111]. In epithelioid hemangioma (EH), FOS or FOSB gene rearrangements are commonly present in bone and soft tissue EHs, although these are rare in cutaneous EH [112]. Epithelioid endothelial cells usually express FBJ murine osteosarcoma viral oncogene homolog B (FOSB) via IHC [113]. Arteriovenous malformations are now recognized as vascular malformations rather than neoplasms; terms such as arteriovenous hemangioma, cirsoid aneurysm, and acral arteriovenous tumor are no longer recommended [114].
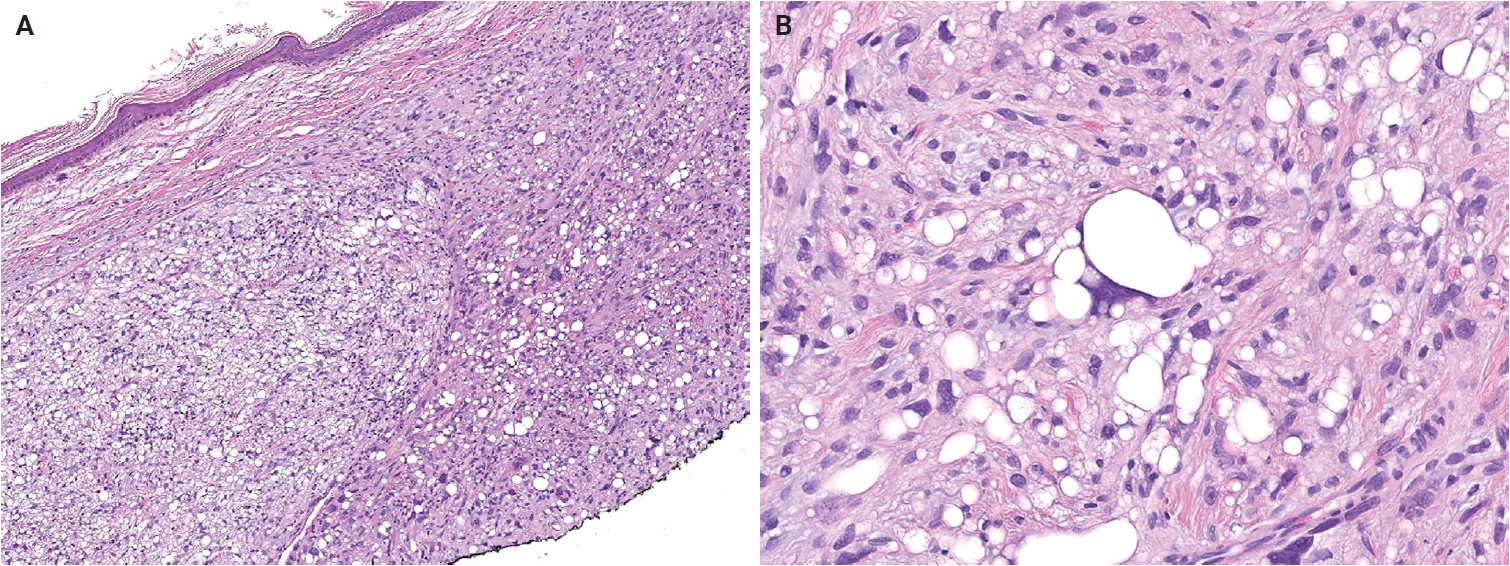
- Pseudomyogenic hemangioendothelioma (PHE) is characterized by recurrent FOSB rearrangements, with various fusion partners such as SERPINE1::FOSB, ACTB::FOSB, WWTR1::FOSB, CLTC::FOSB, and EGFL7::FOSB [115-120], resulting in overexpression of FOSB and constitutive transcriptional activity [120]. Fusion partner type may influence clinical features: tumors with ACTB::FOSB fusions are often solitary, whereas those with EGFL7::FOSB tend to be more aggressive [120]. Histologically, PHE is characterized by fascicles or sheets of spindled to epithelioid cells with vesicular nuclei and eosinophilic cytoplasm and showing expression of ERG and cytokeratin (AE1/AE3) (Fig. 7A, B). In epithelioid hemangioendothelioma (EHE), >90% of cases harbor WWTR1::CAMTA1 fusion resulting from t(1;3)(p36;q25) translocation [121,122], whereas a minor subset exhibits a YAP1::TFE3 fusion [123,124]. Nuclear expression of calmodulin-binding transcription activator 1 (CAMTA1) serves as a surrogate marker for WWTR1::CAMTA1 fusion, and nuclear transcription factor E3 (TFE3) positivity indicates YAP1::TFE3 fusion. Additionally, the loss of expression of the C-terminus of yes-associated protein 1 (YAP1) is specific to tumors with YAP1::TFE3 fusions [125]. In composite hemangioendothelioma (CHE), approximately 27% of tumors harbor YAP1::MAML2 fusions [126,127]. Neuroendocrine CHE is recognized as a distinct subtype characterized by a mixture of retiform hemangioendothelioma-like and EHE-like areas, together with a striking nested component, and by expression of neuroendocrine markers, most commonly synaptophysin (Fig. 8A, B) [126,128]. Subsets of neuroendocrine CHEs harbor PTBP1::MAML2 and EPC1::PCH2 fusions [128,129], and CHEs with YAP1::MAML2 fusions show loss of YAP1 C-terminus expression [125].
- Kaposi sarcoma (KS) is classified into four clinical subtypes: classic, endemic African, acquired immunodeficiency syndrome–associated, and iatrogenic [130]. Genomic profiling reveals recurrent 11q13 gains with amplification of FGF4 and FGF3 (INT2) [131]. Early-stage KS shows clonal loss of the Y chromosome, whereas late (nodular) lesions accumulate copy-number alterations on chromosomes 16, 17, 21, X, and Y [132]. Cutaneous angiosarcoma demonstrates expanded genetic heterogeneity (Fig. 9A, B) [133]. Tumors arising in the head, neck, face, and scalp frequently exhibit ultraviolet (UV) radiation-associated mutation signature and high tumor mutation burden, which may have implications for immuno-oncology-based therapy [134]. MYC amplifications are nearly universal in secondary angiosarcoma (post-radiation or lymphedema-associated) but occur in <10% of primary tumors [135-137]. The most common recurrent mutations involve KDR, PTPRB, and PLCG1 (approximately 40%) [138,139], whereas mutations in RAS, PIK3CA, TP53, FLT4, and TIE1 are less frequent [140,141]. Additionally, a CIC-altered subset occurs in younger patients and is associated with poorer disease-free survival [142].
- Papillary hemangioma
- Papillary hemangioma is a distinctive benign vascular proliferation characterized by dilated vascular channels containing prominent intraluminal papillary projections [143]. It occurs almost exclusively in the head and neck region and predominantly affects adult men [144,145]. Clinically, it presents as a solitary, long-standing, bluish-red papule, measuring up to 11 mm in diameter.
- Histologically, papillary hemangioma demonstrates widely dilated, thin-walled vascular channels in the dermis, containing numerous intraluminal papillary projections. These papillary structures comprise capillary-sized vessels lined by plump endothelial cells and surrounded by pericytes. Endothelial cells often contain intracytoplasmic eosinophilic globules (so-called ‘thanatosomes’), corresponding to dilated lysosomes containing fat vacuoles and cellular debris. Immunohistochemically, endothelial cells are positive for CD31, CD34, and ERG, but negative for lymphatic markers such as podoplanin (D2-40).
- Poikilodermatous plaque-like hemangioma
- PPLH is an acquired benign vascular proliferation characterized by a band-like pattern of small blood vessels in the superficial dermis [146]. Almost all lesions arise on the lower extremities [147,148], predominantly affecting older men. Clinically, PPLH presents as slowly growing, asymptomatic, erythematous to violaceous, scaly plaques, measuring 20−70 mm in diameter, often exhibiting poikilodermatous changes. Lesions are usually solitary but may be multiple. The course is indolent, and lesions tend to persist despite treatment.
- Histologically, PPLH demonstrates a band-like proliferation of small-caliber blood vessels lined by bland endothelial cells within the papillary and superficial reticular dermis. The affected area shows loss or marked reduction of elastic fibers, and a grenz zone is absent. Variable degrees of hyperkeratosis, acanthosis, and mild perivascular lymphocytic infiltration are also commonly observed.
- Acquired elastotic hemangioma
- AEH is a rare, benign vascular proliferation characterized by a superficial, band-like arrangement of small blood vessels in sun-exposed skin [149]. It is typically associated with solar elastosis. AEH predominantly occurs on the forearms or lateral neck and presents as an asymptomatic papule or plaque, primarily affecting middle-aged to elderly adults, with a slight female predominance [150,151]. Multiple lesions are rare, and chronic sun damage is considered the primary etiologic factor.
- Histologically, AEH exhibits a band-like proliferation of capillary-sized vessels, each surrounded by a layer of pericytes and oriented parallel to the epidermis within the upper reticular dermis. These vessels are set against a background of prominent solar elastosis.
- Hobnail hemangioendotheliomas
- PILA and RHE are uncommon, rarely metastasizing vascular neoplasms—both feature hobnail endothelial proliferations with a lymphatic endothelial phenotype [107]. PILA typically arises in the proximal extremities, particularly the buttock and thigh [152-154], whereas RHE predominantly occurs in the distal extremities, especially the lower leg [155,156]. Fewer than 50 cases of PILA and RHE have been reported [152,155,156]. PILA predominantly affects infants, with a slight female predominance, whereas RHE occurs in children or young adults, with no sex predilection. The etiology remains uncertain; some RHE cases are associated with prior radiotherapy [155], lymphedema [155,157], or cystic lymphangioma [158]. A subset of RHE harbors YAP1 rearrangements, most often YAP1::MAML2 fusions [127].
- Histologically, PILA and RHE are morphologically similar, composed of darkly staining hobnail or columnar endothelial cells. PILA exhibits poorly defined proliferation of slit-like or dilated lymphatic channels, occasionally associated with cavernous lymphatic malformation. In contrast, RHE exhibits infiltrative growth of elongated, branching vascular channels resembling the rete testis. Shared features include hyaline fibrosis, intraluminal papillary tufts lined by hobnail endothelial cells—more conspicuous in PILA—and a lymphocytic infiltrate. Immunohistochemically, hobnail endothelial cells of PILA and RHE express pan-endothelial markers (CD31, ERG, Friend leukemia virus integration 1 [FLI1], and CD34) and lymphatic markers (podoplanin [D2-40], vascular endothelial growth factor receptor 3 [VEGFR3], and prospero homeobox 1 [PROX1]) [158,159]. Tumors harboring YAP1 rearrangements show loss of YAP1 C-terminal expression [151]. Focal synaptophysin expression and shared YAP1::MAML2 fusions support a pathogenetic link between HHE and CHE.
VASCULAR TUMORS
- Key updates
- No major revisions or newly recognized entities have been added to this family in the 5th edition. Myofibroma and myofibromatosis, previously categorized among fibroblastic, myofibroblastic, and fibrohistiocytic tumors, have been reclassified as pericytic and perivascular tumors. In glomus tumor, sporadic tumors often show rearrangements involving MIR143 and NOTCH genes [160-162], whereas BRAF p.V600E mutations have been reported in rare malignant cases [163,164]. Demonstration of MIR143::NOTCH1, MIR143::NOTCH2, or MIR143::NOTCH3 fusions can aid in diagnosis [161,162].
- The 5th edition includes newly recognized multicentric and intravascular subtypes of myopericytoma [165]. Pericytoma-like neoplasms harboring recently identified gene fusions, such as ACTB::GLI and SRF::RELA, are likely unrelated to myopericytoma [166,167]. Angioleiomyomas exhibit cytogenetic abnormalities, including monosomy of chromosome 13 and losses of 6p, 21q, and 13q [168,169]; rare cases harbor NOTCH gene fusions [161,162]. In addition to solid, venous, and cavernous subtypes, hybrid subtypes have also been described. Similar to glomus tumors and myopericytomas, angioleiomyomas demonstrate diffuse immunoreactivity for the pericytic marker regulator of G-protein signaling 5 [170].
- Myofibroma and myofibromatosis
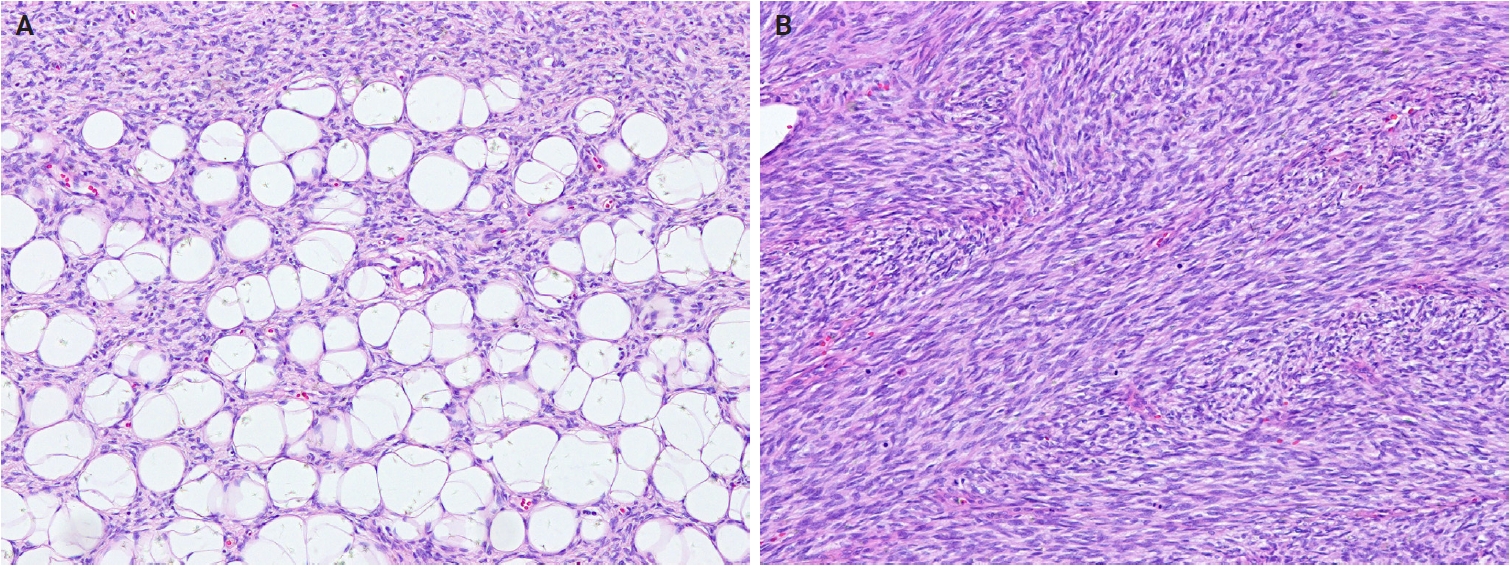
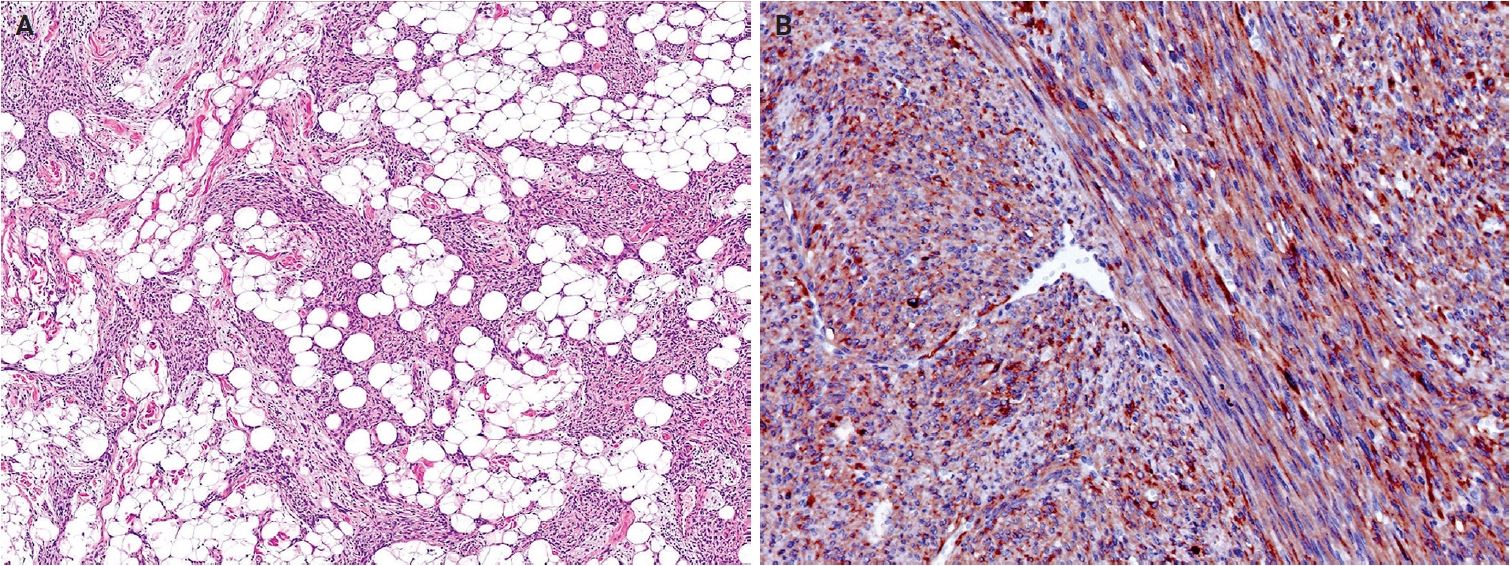
- Myofibroma and myofibromatosis are benign mesenchymal neoplasms of presumed myofibroblastic lineage, presenting as solitary, multicentric, or generalized lesions [171]. Solitary lesions most commonly involve the skin or subcutaneous tissue of the head and neck, upper extremities, or trunk. However, deep lesions involving muscles and bones are uncommon [172]. Multicentric lesions share a similar distribution, whereas generalized lesions involve visceral organs. Solitary lesions predominantly affect males, while multicentric lesions occur mainly in females. Myofibroma accounts for approximately 12% of pediatric soft tissue lesions, with 90% of cases occurring in children and 65% presenting before age 2. Molecularly, familial myofibromatosis exhibits germline PDGFRB mutations (approximately 89%) and NOTCH3 mutations (approximately 11%), whereas sporadic solitary lesions frequently harbor somatic PDGFRB mutations [173,174]. PDGFRB mutation types differ between familial and sporadic myofibromatosis cases, and SRF rearrangements have been reported in highly cellular tumors [175].
- Histologically, myofibroma shows a biphasic growth pattern comprising alternating bundles of primitive, small, darkly staining, rounded cells and plump, spindle-shaped mesenchymal cells (Fig. 10A, B). Tumors are richly vascularized, with numerous thin-walled, hemangiopericytoma-like vessels and mature eosinophilic myoid cells, often accompanied by myointimal nodular proliferations (‘vascular balls’). Foci of calcification and ischemic-type necrosis may be present, and mitotic activity is typically low. Immunohistochemically, tumor cells are diffusely positive for SMA, while caldesmon is negative or focally positive, and desmin is generally negative.
PERICYTIC AND PERIVASCULAR TUMORS
- Key updates
- No major revisions or newly recognized entities have been added to this family in the 5th WHO classification. Smooth muscle hamartoma (SMH) and Epstein-Barr virus–associated smooth muscle tumor (EBV-SMT) are newly included in the family of smooth muscle tumors. In the 5th edition, dermal-confined lesions previously termed ‘cutaneous leiomyosarcoma’ are designated as ‘atypical intradermal smooth muscle neoplasm’, whereas the term ‘cutaneous leiomyosarcoma’ is reserved for tumors with overt subcutaneous fat infiltration (Fig. 11A, B) [176]. Prognosis is excellent; no metastases have been reported in the largest series to date, comprising 84 cases of completely excised dermal-confined tumors [177]. Although data remain limited, dermal tumors with minimal subcutaneous extension have a low risk of recurrence or metastasis. Histological grading does not provide prognostic value.
- Smooth muscle hamartoma
- SMH is a benign dermal lesion composed of haphazardly arranged smooth muscle bundles [178]. It most commonly occurs on the trunk or extremities, with less frequent involvement of the head, neck, and genital regions [179]. A positive pseudo-Darier sign is observed in up to 80% of congenital cases. Most congenital SMHs are solitary, although rare multiple, familial, or diffuse forms have been reported [180]. Diffuse congenital SMHs may be associated with Michelin tire baby syndrome [181], and combined lesions with congenital or blue nevi have also been reported [182,183]. SMH exhibits a slight male predominance, and congenital cases are uncommon, with an estimated incidence of 1 in 2,600 births [182]. Molecularly, a subset of congenital SMHs harbors postzygotic ACTB mutations, also identified in Becker nevi, suggesting a shared biological basis between these lesions [183].
- Histologically, SMH exhibits haphazard bundles of mature smooth muscle within the dermis, often adjacent to pilosebaceous units [184]. Cytological atypia, pleomorphism, and mitotic activity are absent. Focal retraction artifacts may be present around smooth muscle bundles. Lesional cells express smooth muscle markers. The overlying epidermis frequently demonstrates hyperkeratosis, acanthosis, and basal hyperpigmentation, resembling features of a Becker nevus.
- EBV-associated smooth muscle tumor
- EBV-SMT is a rare EBV-driven smooth muscle neoplasm that develops in immunosuppressed patients [185]. The central nervous system is the most commonly affected, followed by the liver, lungs, and subcutaneous tissues of the trunk and extremities [186-189]. EBV-SMT occurs in approximately 1%–5% of immunosuppressed individuals [189], affecting a wide age range, most often adults, with a slight female predominance [190]. Tumorigenesis is driven by EBV type 2 infection in the context of a congenital immunodeficiency disorder, post-transplant immunosuppression, or human immunodeficiency virus infection [189,191]. EBV infects vascular smooth muscle progenitor cells via CD21 receptor-mediated entry or through EBV-infected lymphocytes [192,193]. Aberrant activation of the mTOR/AKT signaling pathway and MYC overexpression contribute to pathogenesis [193,194]. Multiple lesions typically represent independent clonal proliferations rather than metastases [186]. Prognosis is generally favorable and depends more on immune status than on tumor morphology [186,189].
- Histologically, EBV-SMTs are well-circumscribed lesions composed of interlacing fascicles of spindle-shaped myoid cells within a collagenous stroma, irrespective of anatomical site [195,196]. A variable admixture of smaller rounded cells and intratumoral lymphocytes is typically present, and focal myxoid change may be observed. Mitotic activity is generally low, although rare tumors exhibit leiomyosarcoma-like features, including nuclear atypia, necrosis, and increased mitotic activity [190,191]. Immunohistochemically, tumor cells express smooth muscle markers, including SMA, muscle-specific actin, desmin, and caldesmon. EBV infection can be confirmed by in situ hybridization test for Epstein-Barr virus-encoded small RNA.
SMOOTH MUSCLE TUMORS
- Key updates
- In the 5th edition, dermal hyperneury (DHN)/epithelial sheath neuroma (ESN) and hybrid nerve sheath tumor have been newly described within the neural tumors family. In solitary circumscribed neuroma (SCN), the plexiform subtype has been newly recognized as part of the SCN spectrum [197]. Dermal nerve sheath myxoma (DNSM) is now defined as a benign peripheral nerve sheath tumor (Fig. 12A, B) [198]. Pathological and molecular evidence supports its Schwann cell origin [198-201]. Gene expression profiling of DNSM aligns with dermal schwannoma, whereas cellular neurothekeoma exhibits molecular similarities to cellular dermatofibroma (fibrous histiocytoma) [201].
- Three subtypes of perineurioma are recognized: reticular, sclerosing, and malignant [202]. Perineuriomas share pathogenetic features with schwannomas and meningiomas, including 22q12 deletion, monosomies, and NF2 mutations [203-205]. Soft tissue perineuriomas commonly exhibit a 17q11 deletion involving the NF1 locus. The sclerosing subtype demonstrates a distinctive 10q24 rearrangement [206]. Malignant perineuriomas are associated with loss of chromosome 13q and small deletions on chromosomes 3, 6, and 9 [205,207].
- Neurofibroma formally recognizes melanotic neurofibroma as a distinct subtype [208]. Multiple tumors, particularly plexiform and diffuse subtypes, are strongly associated with neurofibromatosis type 1 (NF1) and type 2 (NF2). Genotype–phenotype correlations have been established in NF1 [209]. Loss of CDKN2A at chromosome 9p21.3 may contribute to the development of atypical neurofibromatous tumor of uncertain biological potential [210]. Schwannoma is classified into six subtypes: plexiform, epithelioid, ancient schwannoma, cellular, microcystic/reticular, and neuroblastoma-like [211].
- Granular cell tumor (GCT) is a benign neuroectodermal tumor derived from Schwann cells [212]. Loss-of-function mutations in ATP6AP1 and ATP6AP2, which are mutually exclusive, occur in approximately 70% of cases [213] and are considered pathognomonic of GCT. Malignant GCT is characterized by overt malignant features, including increased cellularity, prominent spindling, a high nuclear-to-cytoplasmic ratio, marked pleomorphism, elevated mitotic activity (including atypical figures), necrosis, and a high Ki-67 proliferation index [214-216].
- Cutaneous malignant peripheral nerve sheath tumor (MPNST) is rare [217]. Unlike deep MPNSTs, cutaneous MPNSTs are less frequently associated with NF1. Conventional deep-seated MPNSTs harbor inactivating mutations in NF1, CDKN2A/CDKN2B, and polycomb repressive complex 2 (PRC2) core components (EED or SUZ12), resulting in complete loss of PRC2 function [218-220]. Approximately 80% of high-grade MPNSTs show PRC2 inactivation, with loss of histone H3 lysine 27 trimethylation (H3K27me3) expression. The loss of H3K27me3 is a valuable diagnostic marker for these tumors [221,222]. In contrast, epithelioid MPNSTs represent a molecularly distinct subtype, characterized by SMARCB1 inactivation in approximately 75% of cases [223]. Mutations in NF1, CDKN2A/CDKN2B, or PRC2 are uncommon in this subtype [218,224].
- Dermal hyperneury/epithelial sheath neuroma
- DHN is a form of small nerve hypertrophy characterized by large, prominent dermal nerve fibers [225]. ESN shows similar histological features, with mature squamous epithelium surrounding the perineurium. DHN/ESN is classified into syndromic and sporadic subtypes. Sporadic DHN shows a slight predilection for the trunk [226], whereas ESN most commonly arises on the back [227]. Sporadic lesions typically present in the seventh decade of life, whereas syndromic mucocutaneous neural proliferations may occur in patients as young as 5 years [228]. Mucocutaneous neural proliferations have been described in multiple endocrine neoplasia type 2B (MEN2B) [229], MEN2A [230], NF2 [231], and Cowden disease [232]. ESN occurs exclusively as a sporadic lesion. Pathogenesis may involve PTEN- and rearranged during transfection (RET)–mediated signaling pathways, particularly in syndromic cases [233].
- Histologically, both DHN and ESN demonstrate aggregated neural tissue with enlarged nerve fibers located within the dermis or at the dermal-subcutaneous junction. Morphology is consistent between syndromic and sporadic cases. Nerve bundles are typically 1.5–4 times larger than normal and may exhibit mild neuroma-like disorganization. Occasionally, perineurial lymphoplasmacytic or histiocytic inflammation and intraneural mucin deposition are observed. ESN exhibits mature squamous epithelium forming an epithelial sheath around enlarged nerve fibers. Subcutaneous extension is rare [234].
- Hybrid nerve sheath tumors
- Hybrid nerve sheath tumors are rare benign peripheral nerve sheath tumors that exhibit combined features of two or more conventional types, most commonly schwannoma, neurofibroma, or perineurioma [235]. They occur across a wide anatomical range, most frequently in the dermis or subcutaneous tissue [236,237]. The most common subtype is hybrid schwannoma/perineurioma, which predominantly arises in the fingers [238]. Hybrid schwannoma/perineurioma usually occurs sporadically, whereas hybrid neurofibroma/schwannoma exhibits a striking association with NF1 and NF2 [239]. A high prevalence of hybrid schwannoma/neurofibroma morphology was found in tumors from patients with schwannomatosis (71%) [239,240]. Hybrid neurofibroma/perineurioma has also been reported in association with NF1 [241,242]. Most hybrid schwannoma/perineuriomas harbor VGLL3 gene rearrangements, supporting their classification as a distinct pathological entity rather than a simple histological overlap [243].
- Histologically, a hybrid schwannoma/perineurioma exhibits an intimate admixture of alternating Schwann cells and perineurial cells. Schwann cells have wavy, tapering nuclei and eosinophilic cytoplasm, whereas perineurial cells have slender nuclei and delicate, elongated cytoplasmic processes. The Schwann cell component is usually predominant and may exhibit degenerative nuclear atypia (‘ancient change’), while mitoses are rare. Hybrid neurofibroma/schwannoma is characterized by schwannomatous nodules or bundles of Schwann cells within an otherwise typical neurofibroma. The neurofibromatous areas contain a mixture of fibroblasts, Schwann cells, and perineurial cells [242,244]. Hybrid neurofibroma/perineurioma shows plexiform neurofibroma with foci of perineurial differentiation, which are often subtle and require IHC for confirmation. Immunohistochemically, Schwann cells are positive for S100 protein and SRY-box transcription factor 10 (SOX10), whereas perineurial cells express epithelial membrane antigen (EMA), claudin-1, and GLUT1, and fibroblasts are positive for CD34.
NEURAL TUMORS
- Key updates
- The 5th edition introduces several newly described entities in the family of tumors of uncertain differentiation, including perivascular epithelioid cell tumor (PEComa), angiomatoid fibrous histiocytoma (AFH), neurotrophic tyrosine receptor kinase (NTRK)–rearranged spindle cell neoplasm (NRSCN), superficial CD34-positive fibroblastic tumor (SCFT), and CRTC1::TRIM11 cutaneous tumor (CTCT). The SCFT, previously classified as a fibroblastic and myofibroblastic tumor in the 5th edition of the WHO classification of soft tissue tumors, has also been reclassified as a tumor of uncertain differentiation.
- Cellular neurothekeoma is a benign cutaneous neoplasm of uncertain histogenesis, distinct from nerve sheath myxoma (previously called myxoid neurothekeoma) [245]. The most consistently expressed markers include CD63 (NKI/C3) [246,247], neuron-specific enolase [248], protein gene product 9.5 (PGP9.5) [249], and S100 calcium-binding protein A6 (S100A6) [250]. The 5th edition expands the immunoprofile to include keratinocyte basal antigen 62 (KBA.62) and preferentially expressed antigen in melanoma (PRAME) [251,252]. Non-neural granular cell tumor is a rare, low-grade neoplasm of mesenchymal cells of unknown lineage, characterized by prominent cytoplasmic granularity [253]. ALK rearrangements have been reported in 60% of cases in a small series [254]. Atypical fibroxanthoma is defined as a dermally confined, low-grade neoplasm of uncertain histogenesis [255]. Genomic alterations include deletions at 9p and 13q, TERT mutations, and a variety of abnormalities similar to pleomorphic dermal sarcoma (PDS) [256-258]. Morphologic patterns include spindle cell, clear cell, granular cell, keloidal, myxoid, sclerotic, and pigmented forms [259-261].
- PDS is a rare tumor affecting older adults with chronically UV radiation-exposed skin [262]. It is characterized by a high UV-related mutation burden, including mutations in TP53, NOTCH family genes, the TERT promoter, and alterations in CDKN2A and CDKN2B [256,258]. This mutation profile parallels that of cutaneous squamous cell carcinoma. Immunohistochemically, PDS commonly expresses CD10, CD99, and platelet-derived growth factor receptor beta (PDGFRB).
- Epithelioid sarcoma (ES) is a malignant soft tissue tumor that exhibits hybrid mesenchymal and epithelial morphology and immunophenotypes [263]. Loss of nuclear SMARCB1 (INI1) expression is characteristic of ES; however, a subset retains SMARCB1 (INI1) and instead shows aberrant expression of other SWI/SNF complex components, including SMARCA4 (BRG1), SMARCC1 (BAF155), and SMARCC2 (BAF170) [264]. Genomic analyses have revealed complex copy-number aberrations and a relatively high mutational burden [265].
- Dermal clear cell sarcoma (CCS) is a malignant neoplasm of uncertain histogenesis with melanocytic differentiation, defined by EWSR1::ATF1 or EWSR1::CREB1 gene fusion [266]. Approximately 70%–90% of CCSs harbor an EWSR1::ATF1 fusion resulting from t(12;22)(q13;q12). Identification of EWSR1 rearrangement and/or confirmation of these fusions aids diagnosis, particularly in distinguishing CCS from malignant melanoma. In contrast to malignant melanomas, PRAME expression in CCS is typically absent or limited to focal areas [267,268].
- Ewing sarcoma is a small round cell sarcoma defined by gene fusions involving a FUS-EWSR1-TAF15 (FET) family gene, most commonly EWSR1, and an erythroblast transformation-specific (ETS) family transcription factor [269]. All cases harbor FET::ETS fusions, which constitute the defining molecular hallmark. The tumorigenesis of cutaneous Ewing sarcoma is driven by oncogenic transcription factors encoded by chimeric FET::ETS fusion genes, most frequently EWSR1::FLI1 (>95%), and, less commonly, EWSR1::ERG [270-272].
- Epithelioid fibrous histiocytoma
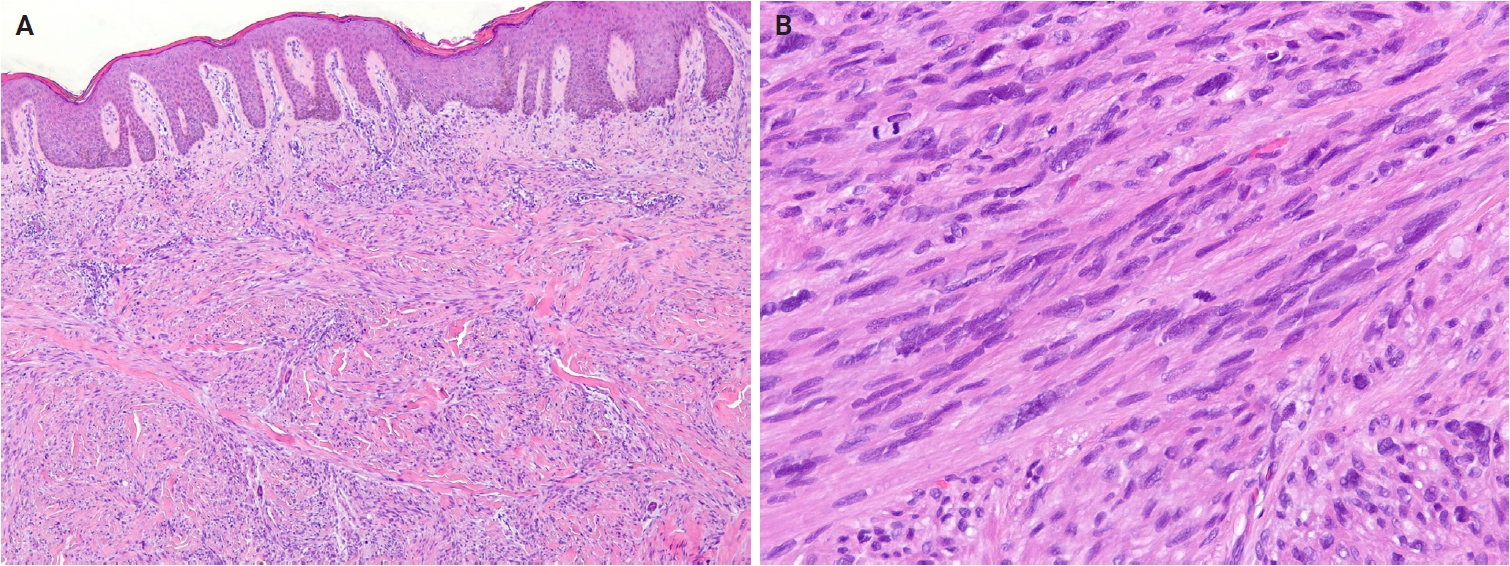
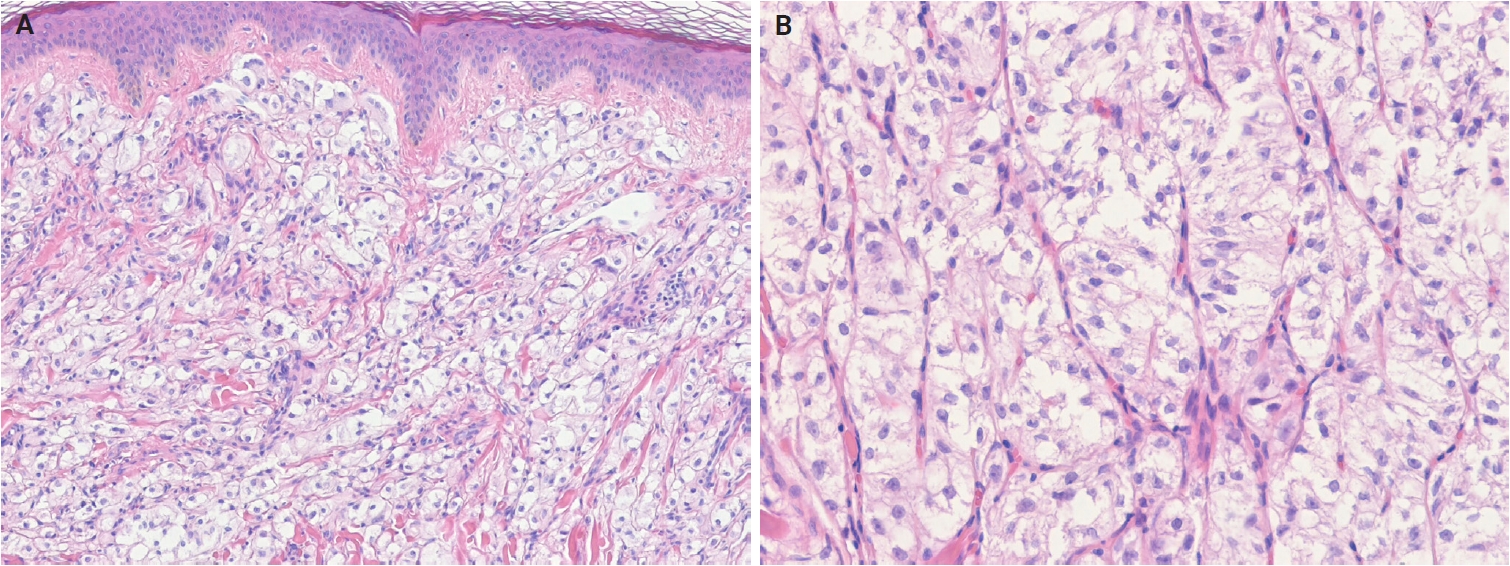
- EFH is a distinctive, benign cutaneous neoplasm composed of epithelioid cells [273]. It most commonly arises in the lower extremities and, less frequently, in the upper extremities, trunk, or head and neck [274,275]. EFH typically occurs in young to middle-aged adults (median age, 40 years), with a female predominance. Molecularly, approximately 90% of EFHs harbor ALK rearrangements, while a small subset shows mutually exclusive PRKC gene fusions [276,277]. SQSTM1 and VCL are the most common ALK fusion partners, whereas DCTN1, ETV6, PPFIBP1, SPECC1L, TPM3, PRKAR2A, MLPH, CLTC, and EML4 represent rare fusion partners [278-281].
- Histologically, EFH is typically well-circumscribed, and exophytic lesions often exhibit an adnexal collarette (Fig. 13A). The tumor consists of uniform epithelioid cells with vesicular nuclei, small nucleoli, and pale eosinophilic or amphophilic cytoplasm (Fig. 13B). Binucleated forms are frequently observed. Thin-walled vessels are common, often with perivascular accentuation of tumor cells. Morphological variants include chondroblastoma-like pericellular calcification [279,282] and spindle cell-predominant forms [283,284]. Immunohistochemically, ALK is overexpressed in approximately 90% of cases [285], with D5F3 or 5A4 clones demonstrating higher sensitivity than the ALK1 clone [276,286]. EMA is positive in approximately 65% of cases [286], and CD30 expression may also be present.
- PEComa
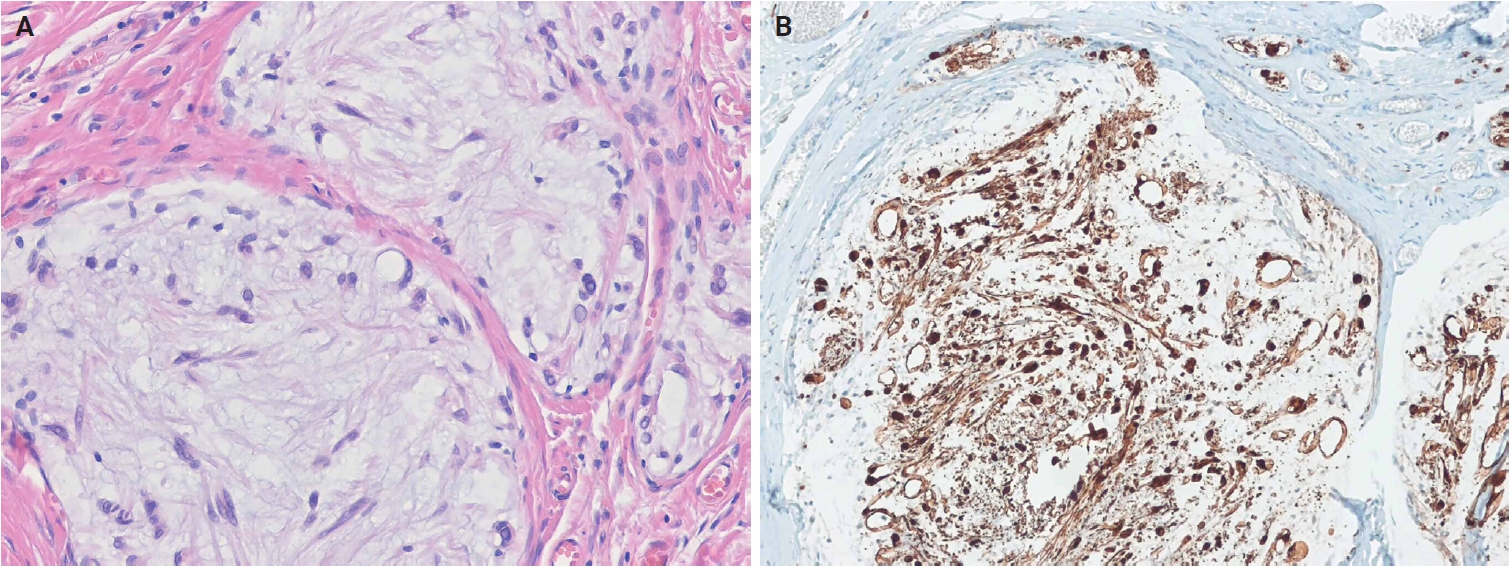
- PEComas are mesenchymal neoplasms composed of distinctive perivascular epithelioid cells that coexpress melanocytic and smooth muscle markers [287]. Cutaneous PEComas are rare and most commonly involve the lower extremities, with less frequent occurrence on the trunk, head, or neck [288-290]. They affect individuals across a wide age range (15–81 years), with a peak incidence in middle age, and show a female predominance [288,289]. Most cases are sporadic; however, fibroma-like cutaneous PEComa associated with tuberous sclerosis has been reported [291]. Molecularly, deletion of 16p involving the TSC2 gene is frequent, and loss of heterozygosity at the TSC2 locus is seen in both sporadic and syndromic tumors [292]. TP53 mutations occur in approximately 63% of TSC2-mutated PEComas [293].
- Histologically, PEComa is composed of epithelioid cells with granular, pale eosinophilic, or clear cytoplasm and round nuclei with small nucleoli (Fig. 14A, B). Most PEComas exhibit a nested or trabecular architecture with a distinctive perivascular growth pattern. Mixed epithelioid-spindle cell morphology and scattered multinucleated cells may occur; however, mitoses are generally rare. Most reported cases of cutaneous PEComa lack malignant features, such as marked pleomorphism, high mitotic activity, and necrosis [292-296]. Malignant primary cutaneous PEComas are exceedingly rare [290,297]. Currently, cutaneous angiomyolipomas are classified as hamartomatous or lipomatous neoplasms with smooth muscle elements, rather than true PEComas. Immunohistochemically, PEComas coexpress melanocytic markers (e.g., human melanoma black 45 [HMB-45], melan-A (MART1), microphthalmia-associated transcription factor [MITF]) and muscle markers (e.g., SMA, desmin, caldesmon) [298,299].
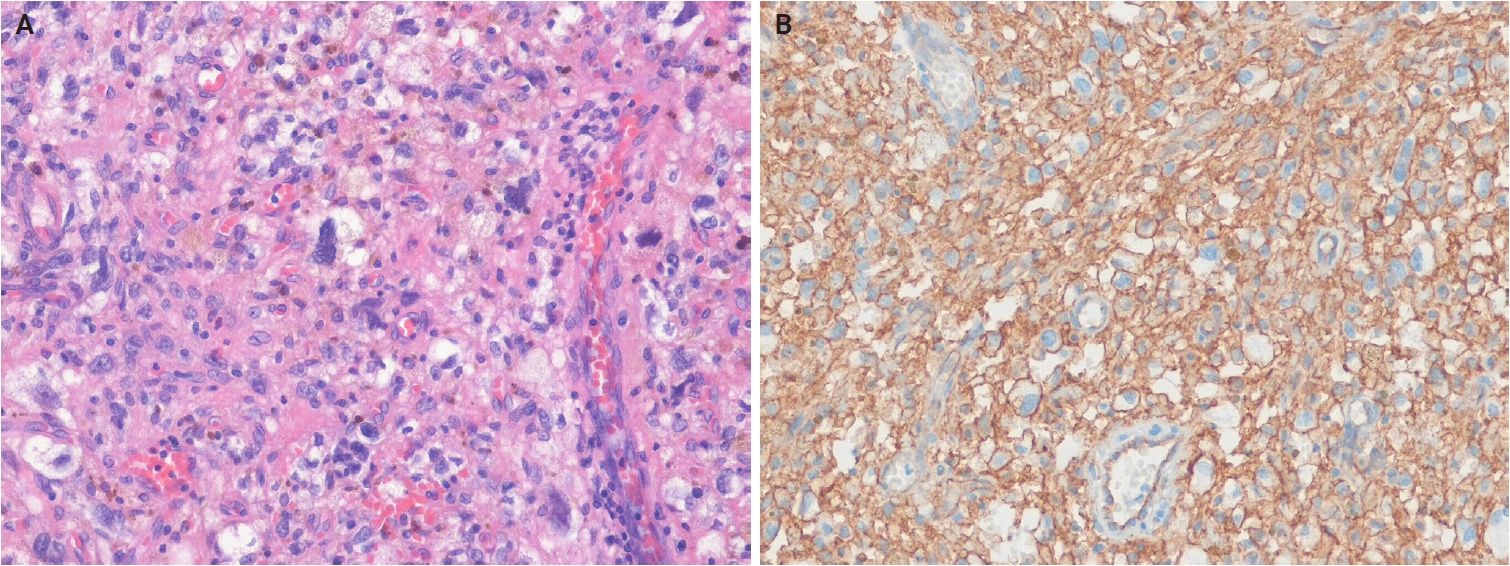
- Angiomatoid fibrous histiocytoma
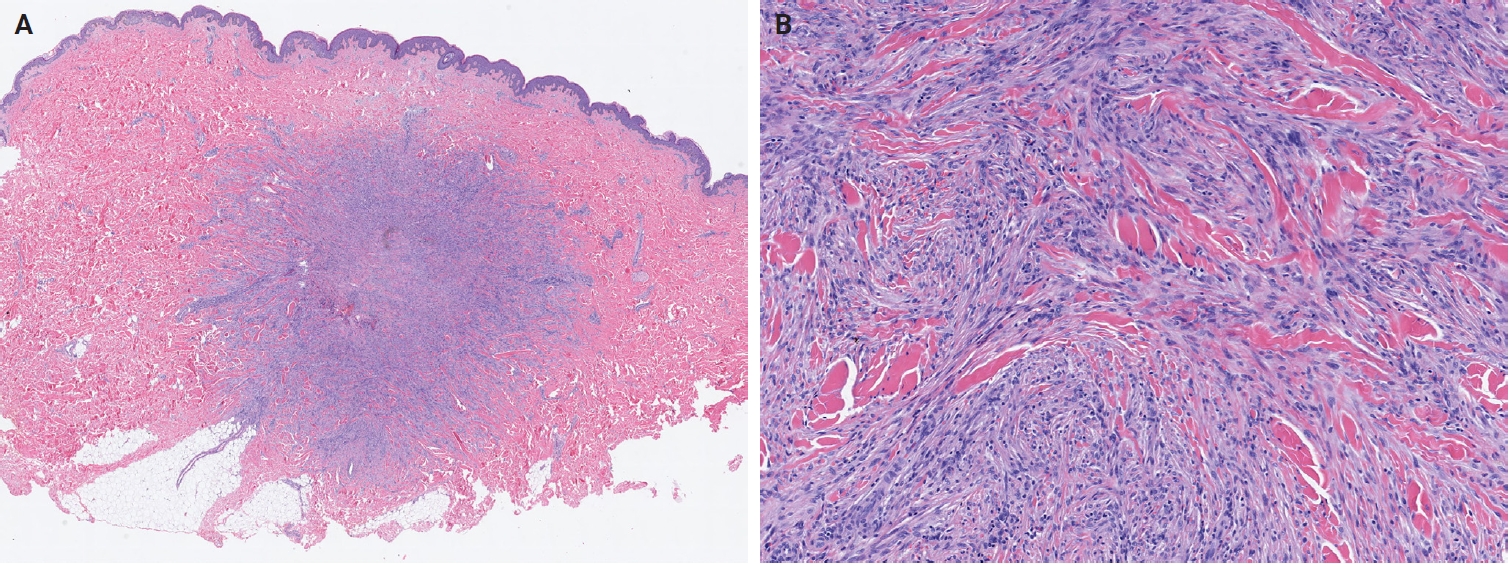
- AFH is a rare soft tissue neoplasm of intermediate malignant potential and uncertain differentiation [300]. It accounts for approximately 0.3% of all soft tissue tumors. It typically presents as a dermal or subcutaneous lesion, most commonly in the extremities, followed by the trunk, head, and neck regions. Approximately two-thirds of cases arise in sites where lymph nodes are normally present [301]. AFH affects a wide age range, but most commonly presents in the first two decades of life [302-304] and shows no sex predilection. Molecularly, the characteristic alteration is t(2;22)(q33;q12), resulting in an EWSR1::CREB1 fusion, followed by t(12;22)(q12;q12), resulting in an EWSR1::ATF1 fusion [305,306]. Rare cases harbor FUS::ATF1 fusion [307,308].
- Histologically, AFH consists of fascicles or sheets of oval to spindle-shaped cells with bland vesicular nuclei and prominent pseudoangiomatous blood-filled spaces. The tumor is characterized by a fibrous pseudocapsule with hemosiderin deposition and a pericapsular rim of lymphoplasmacytic infiltrates containing germinal centers, which may mimic a metastatic lymph node. Mitotic figures are usually infrequent, although atypical mitoses and pleomorphic cells may be present [309,310]. Two histological subtypes have been described: a small round cell subtype resembling undifferentiated round cell sarcoma [301,311] and a myxoid subtype [312,313]. Immunohistochemically, most AFHs express SRY-box transcription factor 9 (SOX9) [314], and approximately 50% are positive for desmin. EMA, CD99, and CD68 demonstrate variable immunoreactivity [301]. The Ki-67 proliferation index is typically low [313].
- NTRK-rearranged spindle cell neoplasm
- NRSCN is a recently recognized molecularly defined spindle cell tumor characterized by activating NTRK or other kinase fusions [315]. These tumors typically arise in the deep dermis and subcutis of the extremities, trunk, and head and neck area [316-318]. Although most cases occur in children and young adults, the age range is wide (0–77 years; median age, 19 years), with no sex predilection [318,319]. Oncogenesis is driven by mitogen-activated protein kinase pathway activation via kinase fusions, most commonly involving LMNA::NTRK1, TPR::NTRK1, or TPM3::NTRK1 fusions [315,316]. Alternative rearrangements involving NTRK2, NTRK3, RAF1, BRAF, RET, MET, ROS1, or ALK have also been reported [318]. NRSCNs are locally infiltrative and prone to local recurrence after incomplete excision. However, metastasis is rare, and the prognostic relevance of histologic grade remains uncertain [318,319].
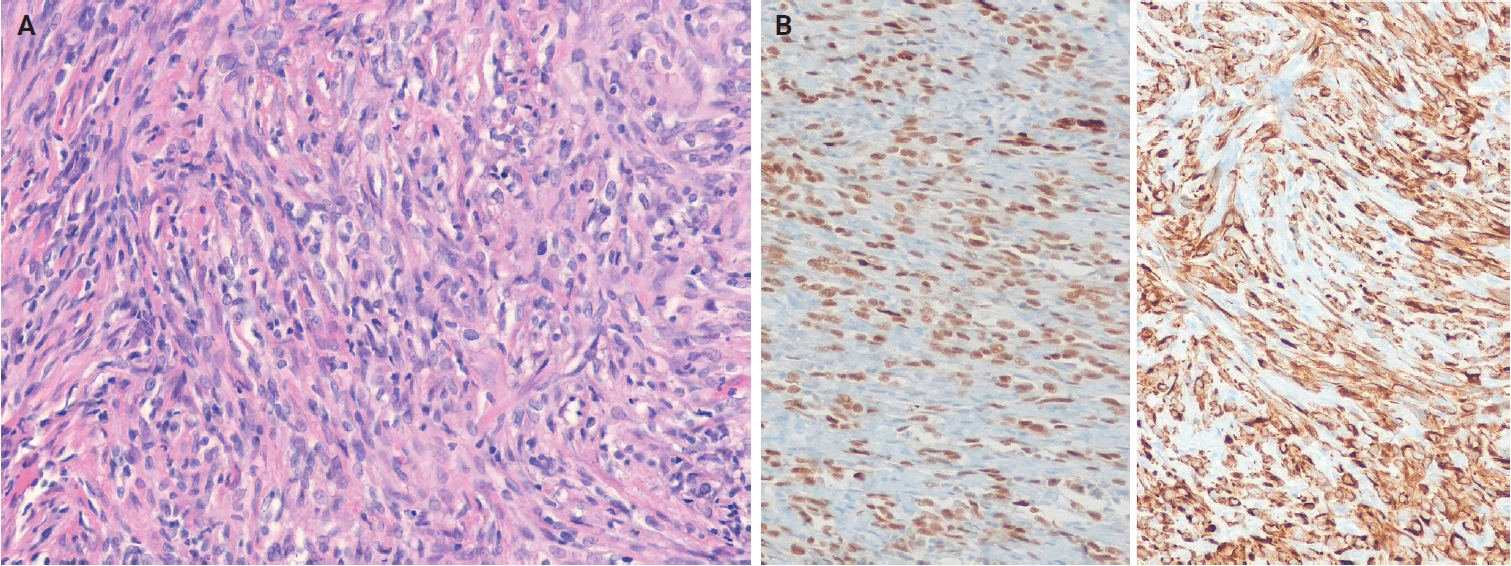
- Histologically, NRSCNs exhibit a broad morphological spectrum. Superficial lesions often show a lipofibromatosis-like pattern, characterized by fibrous septa and spindle cells with ovoid to tapering nuclei and indistinct cytoplasm extending through the adipose tissue (Fig. 15A). Some tumors may be more cellular, with spindled to ovoid cells arranged in fascicles or dispersed loosely in a myxoid stroma. Tumors commonly exhibit hybrid patterns, with highly cellular areas devoid of entrapped adipose tissue admixed with adipose-rich regions. Additionally, lymphocytic infiltrates, staghorn-like vessels, perivascular hyalinization, and stromal collagen bands may also be present. High-grade features such as marked atypia, high mitotic activity, or necrosis are infrequently observed [318,319]. Immunohistochemically, NRSCNs typically coexpress CD34 and S100 protein, with occasional SMA positivity. Tumors with activating NTRK fusions show diffuse pan–tropomyosin receptor kinase (pan-TRK) expression (Fig. 15B) [319,320], but the immunoprofile may be non-specific [316,318]. Therefore, molecular testing is necessary for a definitive diagnosis and for selecting targeted therapy.
- Superficial CD34-positive fibroblastic tumor
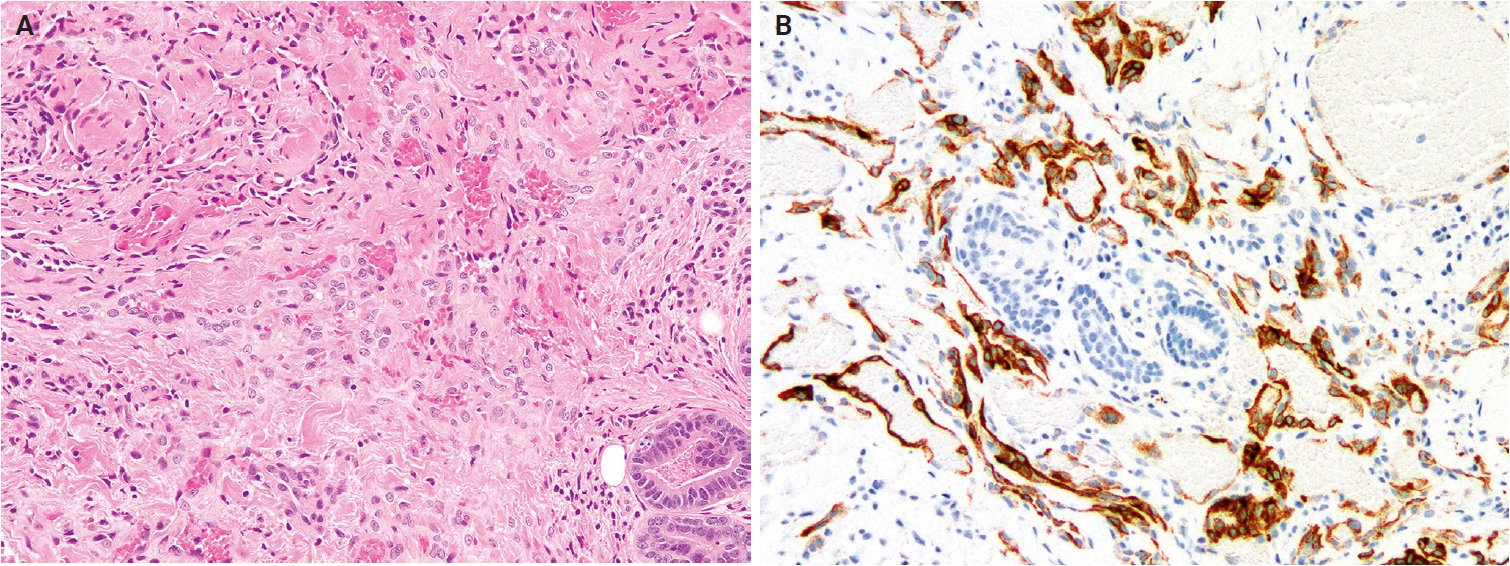
- SCFT is a distinctive, low-grade neoplasm of the skin and subcutaneous tissue, characterized by a consistent morphology and diffuse CD34 expression [321]. It predominantly arises in the lower extremities, particularly the thigh, and less frequently, in the arm, buttock, shoulder, and vulva [322-325]. SCFT typically affects middle-aged adults (median age, 38 years) and shows a slight male predominance [324-327]. Morphological, immunophenotypic, and molecular findings are identical to those of ‘PRDM10-rearranged soft tissue tumor’, supporting their classification as a single entity [325,328]. However, the term ‘SCFT’ is retained because not all tumors demonstrate detectable PRDM10 fusions. PRDM10 rearrangements have been identified in approximately 60% of cases previously classified as SCFT or PRDM10-rearranged soft tissue tumors [325,328]. The prognosis is excellent [323,324].
- Histologically, SCFTs are relatively well-circumscribed but partially infiltrative, composed of spindled to pleomorphic cells arranged in fascicles or sheets (Fig. 16A). Tumor cells have abundant eosinophilic cytoplasm, often granular or glassy, and lipidized cells are frequently observed. Nuclear pleomorphism ranges from moderate to marked, with bizarre, hyperchromatic nuclei, prominent nucleoli, and occasional intranuclear cytoplasmic pseudoinclusions. Mitotic activity is low, and necrosis is rare [322,323]. Mixed inflammatory infiltrates are common, and myxoid stroma or metaplastic bone may rarely be present [324]. Immunohistochemically, SCFTs exhibit diffuse CD34 expression and focal cytokeratin (AE1/AE3) positivity in approximately 70% of cases (Fig. 16B). The Ki-67 proliferation index is low (<5%) [324,325]. PRDM10 is consistently expressed [325], and CADM3 (SynCam3) expression has been reported in 95%–100% of cases [324,325].
- CRTC1::TRIM11 cutaneous tumors
- CTCT is a dermal neoplasm composed of spindle and epithelioid cells with partial melanocytic differentiation, defined by the characteristic CRTC1::TRIM11 fusion [329]. Recently recognized MITF pathway–activated tumors with melanocytic differentiation include three neoplasms defined by ACT::MITF, MITF::CREM, and CRTC1::TRIM11 fusions. In the current WHO classification, ACT::MITF- and MITF::CREM-rearranged tumors are grouped as MITF pathway–activated melanocytic tumors within the melanocytic neoplasms family [330], whereas CTCTs are classified as soft tissue tumors of uncertain differentiation. CTCTs arise at various anatomical sites, most commonly in the extremities (upper, 40%; lower, 30%), followed by the trunk (15%) and, less frequently, the head or mucosal regions (oral and nasal) [331,332]. Approximately 50 cases of CTCT have been reported, with no sex predilection and an age range of 11–87 years (median age, 44 years) [331-338]. The molecular role of CRTC1::TRIM11 fusion protein remains unclear; CRTC1 likely contributes to cell proliferation and differentiation by activating cAMP response element-binding protein 1 (CREB1), which subsequently upregulates MITF expression [339-341]. Clinically, CTCTs are usually indolent; however, local recurrence or metastasis occurs in approximately 10% of cases after excision. Confirmation of the CRTC1::TRIM11 fusion is required for diagnosis.
- Histologically, CTCTs are well-circumscribed, unencapsulated, dermal-based nodules that lack pigment and may resemble CCS. Tumors are composed of nests and intersecting fascicles of spindle to epithelioid cells with vesicular nuclei, prominent nucleoli, and abundant pale cytoplasm (Fig. 17A). Mitoses are present, but infrequent, and epidermal involvement is rare. Immunohistochemically, CTCTs show diffuse SOX10 positivity (Fig. 17B). Approximately 50% of cases exhibit diffuse S100 protein staining, while melan-A (MART1) and HMB-45 display focal expression. Pan-TRK expression is present in approximately 60% of cases, and TRIM11 shows nuclear positivity at the tumor periphery [335-337]. The differential diagnoses include CCS and metastatic malignant melanoma, and definitive diagnosis requires confirmation of CRTC1::TRIM11 fusion.
TUMORS OF UNCERTAIN DIFFERENTIATION
- This review summarizes key updates and newly recognized entities of cutaneous soft tissue tumors included in the 5th edition of the WHO classification of skin tumors. This edition incorporates recent advances in the taxonomy of cutaneous soft tissue tumors and deepens understanding of their pathogenesis by integrating histological and molecular findings. Accurate diagnosis of cutaneous soft tissue tumors requires careful correlation of clinical, microscopic, immunohistochemical, and molecular findings, together with strict adherence to well-defined diagnostic criteria. Further studies are warranted to elucidate molecular pathogenesis and refine the classification of previously unrecognized cutaneous neoplasms.
CONCLUSION
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Conflicts of Interest
J.H.C., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article.
Funding Statement
No funding to declare.
| Tumor family | Biological potential | Tumor type | Clinical features | Histological features | Immunohistochemistry | Molecular features |
|---|---|---|---|---|---|---|
| Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Benign | Fibrous papule | Middle-aged adults; sporadic in most cases; multiple early childhood lesions in tuberous sclerosis; nose, central face | Dome-shaped lesion, with dilated dermal blood vessels; spindle-shaped or stellate fibroblasts; fibrotic or collagenized stroma | CD34 (+); factor XIIIa (+) | Germline mutations in TSC1 or TSC2 (tuberous sclerosis-associated cases) |
| Benign | Fibroblastic connective tissue nevus | Young age (median, 10 years); slight female predominance; trunk, head and neck, limbs | Bland spindle-shaped and ovoid cells arranged in short, intersecting fascicles; entrapped and preserved skin appendages | CD34 (+); SMA (+) | KHDRBS1::NTRK3 (1 case) | |
| Benign | Fibro-osseous tumor of digit | Young adults; rapidly growing; subcutaneous tissues of the proximal phalanx (fingers and toes) | Spindle cells are arranged randomly or in short intersecting fascicles; unmineralized woven bone; vascular or myxoid stroma | SMA (+) in myofibroblasts; SATB2 (+) in osteoblast | USP6 rearrangement | |
| Benign | Inclusion body fibromatosis | Most cases <3 years; ~30% present at birth; dorsal/dorsolateral distal or middle phalanges of 2nd–4th digits (hands and feet) | Spindled cells with bland nuclei and light eosinophilic cytoplasm; a rounded, pale pink, intracytoplasmic inclusion | SMA (+); calponin (+); desmin (+); inclusions: calponin-1 (+), caldesmon (+) | - | |
| Benign | Multinucleate cell angiohistiocytoma | Middle-aged adults; solitary or multiple; dorsal hands, fingers, lower extremities, trunk, back, rarely head and neck | Triad of multinucleated and angulated stromal cells, dermal fibrosis parallel to the epidermis, and increased small-caliber vessels | CD68 (±); factor XIIIa (+) | - | |
| Benign | EWSR1::SMAD3-rearranged fibroblastic tumor | Wide age range (1–68 years; median, 39 years); female predominance; acral sites, most commonly the hands and feet | Hyalinized acellular center with peripheral zones composed of intersecting fascicles of fibroblastic spindle cells; calcification | ERG (+); CD34 (–); SMA (–) | EWSR1::SMAD3 | |
| Vascular tumors | Benign | Papillary hemangioma | Adult men; exclusively head and neck | Dilated, thin-walled dermal vascular channels with numerous luminal papillary projections; intracytoplasmic eosinophilic globules | CD31 (+); CD34 (+); ERG (+) | - |
| Benign | Poikilodermatous plaque-like hemangioma | Elderly men; lower extremities; usually solitary, rarely multiple plaques | Band-like proliferation of small vessels in the superficial dermis; no grenz zone; loss or reduction of elastic fibers | CD31 (+); CD34 (+); ERG (+) | - | |
| Benign | Acquired elastotic hemangioma | Middle-aged to elderly adults; slight female predominance; forearm, lateral neck; associated with chronic sun damage | Band-like proliferation of capillaries surrounded by pericytes; arranged parallel to the reticular dermis, in a background of solar elastosis | CD31 (+); CD34 (+); ERG (+) | - | |
| Intermediate | Hobnail hemangioendothelioma | PILA: infant, proximal extremities, buttock, or thigh; RHE: children or young adults, distal extremities | Hobnail endothelial cells, PILA: poorly defined slit-like or dilated lymphatic channels; RHE: infiltrative, branching vascular channels | Endothelial markers (CD31, ERG, FLI1) (+); lymphatic endothelial markers (podoplanin [D2-40], VEGFR3, PROX1) (+); loss of YAP1 C-terminus expressiona; synaptophysin (±)b | PILA: unknown; RHE: YAP1 rearrangements, most commonly YAP1::MAML2 (subset) | |
| Pericytic and perivascular tumors | Benign | Myofibroma and myofibromatosis | Most cases are in children; sporadic or familial; skin or subcutaneous tissue of the head and neck, upper extremities, trunk | Biphasic pattern of small primitive rounded cells and plump spindled myoid cells; hemangiopericytoma-like vessels | SMA (+); caldesmon (±); desmin (–) | Germline PDGFRB and NOTCH3 mutations (familial); somatic PDGFRB mutations (sporadic); SRF rearrangement (cellular cases) |
| Smooth muscle tumors | Benign | Smooth muscle hamartoma | Broad age spectrum; usually solitary; rare multiple or familial; trunk, extremities, head and neck, external genitalia | Haphazardly arranged bundles of mature smooth muscle in the dermis, often near pilosebaceous units | SMA (+); desmin (+); caldesmon (+) | ACTB mutations (subset) |
| Intermediate | EBV-associated smooth muscle tumor | Wide age distribution; immunosuppressed patients; CNS, liver, lung, subcutaneous tissues of the limbs and trunk | Interlacing fascicles of spindled myoid cells; admixture of smaller rounded cells; intratumoral lymphocytes | SMA (+); desmin (+); caldesmon (+); EBER in situ hybridization (+) | MYC overexpression (subset) | |
| Neural tumors | Benign | Dermal hyperneury/epithelial sheath neuroma | Syndromic cases in children; sporadic cases usually present in older adults; female predominance; lips, eyelid, tongue, trunk | Dermal hyperneury: enlarged, prominent dermal neural fibers; epithelial sheath neuroma: enlarged nerve fibers ensheathed by squamous epithelium | S100 protein (+); neurofilament (+); CD57 (+) in nerve fibers; CKs in perineural epithelial sheaths | - |
| Benign | Hybrid nerve sheath tumors | Any age (peak in young adults); wide anatomic sites; HSP is sporadic; HSN is associated with NF1 and NF2; HNP is associated with NF1 | HSP: admixture of Schwann and perineurial cells; HSN: schwannomatous nodules or Schwann cell bundles within neurofibroma; HNP: plexiform neurofibroma with areas of perineurial differentiation | S100 protein (+) and SOX10 (+) in Schwann cells; EMA (+), claudin-1 (+), and GLUT1 (+) in perineurial cells; CD34 (+) in fibroblasts | VGLL3 rearrangements (most HSPs) | |
| Tumors of uncertain differentiation | Benign | Epithelioid fibrous histiocytoma | Young to middle-aged adults; female predominance; extremities, trunk, head and neck | Exophytic lesions with adnexal collarette; uniform epithelioid cells with abundant eosinophilic to amphophilic cytoplasm | ALK (+) (~90%); EMA (+) (65%); CD30 (±) | ALK rearrangements (~90%); PRKC rearrangements (rare) |
| Benign | PEComa | Wide age (15–81 years); peak incidence in middle age; female predominance; extremities (lower limbs), trunk, head and neck | Epithelioid cells with granular, eosinophilic to clear cytoplasm; nested or trabecular architecture; perivascular growth; rare malignant features (mitotic activity, necrosis, and pleomorphism) | Melanocytic markers (HMB-45, melan-A [MART1], MITF) (+); smooth muscle markers (SMA, desmin, caldesmon) (+) | Frequent deletion of 16p; loss of heterozygosity at TSC2; TP53 mutations (63% of TSC2-mutated tumors) | |
| Intermediate | Angiomatoid fibrous histiocytoma | Wide age range; peak in the first two decades of life; extremities, trunk, head and neck | Fascicles or sheets of ovoid and spindled cells; pseudoangiomatoid spaces; peripheral cuff of lymphoplasmacytic cells | SOX9 (+); desmin (+) (~50%); EMA (±); CD99 (±); CD68 (±) | EWSR1::CREB1 (most frequent); EWSR1::ATF1, FUS::ATF1 (rare) | |
| Intermediate | NTRK-rearranged spindle cell neoplasm | Any age, most commonly pediatric; no sex predilection; extremities, trunk, head and neck | Haphazardly arranged spindled cells; infiltrative growth within fat resembling lipofibromatosis; distinctive stromal and perivascular keloidal collagen | CD34 (+); S100 protein (+); pan-TRK (+); SMA (±); SOX10 (–) | NTRK1 rearrangements: LMNA::NTRK1, TPR::NTRK1, or TPM3::NTRK1; others: NTRK2, NTRK3 rearrangements | |
| Intermediate | Superficial CD34-positive fibroblastic tumor | Middle-aged adults; slight male predominance; lower extremities (especially thigh), arm, buttock, shoulder, vulva | Spindle and pleomorphic cells with abundant, granular to glassy cytoplasm; marked nuclear pleomorphism; low mitotic activity | CD34 (+); PRDM10 (+); CADM3 (SynCAM3) (+); CKs (+) (~70%) | PRDM10 rearrangement (~60%) | |
| Malignant | CRTC1::TRIM11 cutaneous tumor | Broad age range (median, 44 years); extremities, trunk, head; oral and nasal mucosa (rare); slow-growing, skin-colored nodule; generally no pigment | Dermal-based, circumscribed; epithelioid to fusiform cells with vesicular nuclei, prominent nucleoli, and pale cytoplasm; intersecting fascicles; | Melanocytic markers (SOX10, S100 protein, melan-A [MART1]) (+)c; TRIM11 (+); Pan-TRK (+) (~60%) | CRTC1::TRIM11 |
WHO, World Health Organization; SMA, smooth muscle actin; SATB2, special AT-rich sequence-binding protein 2; ERG, ETS-related gene; PILA, papillary intralymphatic angioendothelioma; RHE, retiform hemangioendothelioma; FLI1, Friend leukemia virus integration 1; VEGFR3, vascular endothelial growth factor receptor 3; PROX1, prospero homeobox 1; YAP1 C-terminus, yes-associated protein 1 C-terminus; EBV, Epstein-Barr virus; CNS, central nervous system; EBER, Epstein-Barr virus-encoded small RNAs; CK, cytokeratin; HSP, hybrid schwannoma/perineurioma; HSN, hybrid schwannoma/neurofibroma; NF1, neurofibromatosis type 1; NF2, neurofibromatosis type 2; HNP, hybrid neurofibroma/perineurioma; SOX10, SRY-box transcription factor 10; EMA, epithelial membrane antigen; GLUT1, glucose transporter 1; ALK, anaplastic lymphoma kinase; PEComa, perivascular epithelioid cell tumor; HMB-45, human melanoma black 45; MART1, melanoma antigen recognized by T cells 1; MITF, microphthalmia-associated transcription factor; SOX9, SRY-box transcription factor 9; NTRK, neurotrophic tyrosine receptor kinase; pan-TRK, pan–tropomyosin receptor kinase; PRDM10, PR domain–containing protein 10; CADM3, cell adhesion molecule 3; SynCAM3, synaptic cell adhesion molecule 3; TRIM11, tripartite motif–containing 11; +, positive staining; ±, variable staining; –, negative staining.
aLoss of expression of YAP1 C-terminus is seen in subsets of RHE with YAP1 rearrangements;
bLimited synaptophysin expression may be seen in PILA and RHE;
cAll tumors show diffuse SOX10 expression, whereas diffuse S100 protein staining is present in only about 50% of cases and melan-A (MART1) and HMB-45 are expressed only focally.
| Tumor category | Tumor type | Molecular genetic alterations | Immunohistochemistry |
|---|---|---|---|
| Adipocytic tumors | Naevus lipomatosus superficialis | Deletion of 2p24 (1 case) | - |
| Lipoma | Chromosome 12q13-15 rearrangements involving HMGA2; chromosome 16p21 rearrangements involving HMGA1 | HMGA2 (+) | |
| Angiolipoma | Mutations in PRKD2 and PIK3CA | - | |
| Spindle cell/pleomorphic lipoma | Partial or complete loss of chromosome 13q14 (RB1 locus) and/or 16q deletions | Loss of RB1 expression; CD34 (+) | |
| Atypical lipomatous tumor | Amplification of MDM2, CDK4, TSPAN31 (SAS), HMGA2, and CPM at 12q13-q15 region | MDM2 (+); CDK4 (+) | |
| Pleomorphic liposarcoma | Frequent loss of TP53, RB1, and NF1; frequent gain in 1p21, 1q21-q22, 5p13-p15, and 7q22 | - | |
| Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Fibroma of tendon sheath | USP6 fusion with various partners (PKM, RCC1, ASPN, COL1A1, COL3A1, MYH9, MIR22HG, CTNNB1, SPARC, CAP1, EMP1, CYTOR, NR1D1, RAB1A, and TNC) | - |
| Calcifying aponeurotic fibroma | FN1::EGF | EGF overexpression | |
| Sclerotic fibroma | Loss of PTEN (Cowden syndrome) | CD34 (+); loss of PTEN expression (Cowden syndrome) | |
| Nuchal-type fibroma | APC mutations (a few cases); MUTYH mutation (1 case) | May show β-catenin nuclear expression | |
| Gardner fibroma | Germline APC mutations (FAP-associated tumors) | CD34 (+); β-catenin nuclear expression (FAP-associated tumors) | |
| Pleomorphic fibroma | RB1 deletion; germline mutations in TP53 (Li-Fraumeni syndrome) | Loss of RB1 expression; CD34 (+) | |
| Elastofibroma | Copy gains (Xq); alterations to chromosome 1; t(8;12)(q22;q24.3); t(2;19) and t(X;1) rearrangements (some cases) | - | |
| Desmoplastic fibroblastoma | t(2;11)(q31;q12); t(11;17)(q12;p11.2) | FOSL1 overexpression | |
| Dermatofibroma (fibrous histiocytoma) | Recurrent gene fusions involving PKC isoforms (PRKCA, PRKCB, and PRKCD) with various membrane-associated partners (LAMTOR1, PDPN, CD63, and KIRREL family genes) (minority of cases) | SMA (+); desmin (+) (~30%); CD34 (+) (5%) | |
| Superficial fibromatosis | Lack of CTNNB1 and APC mutations | β-catenin nuclear expression (rare cases); SMA (±); desmin (±) | |
| Plexiform fibrohistiocytic tumor | 46,X,del(X)(q13)[3]/46,XX[23], 46,XY,t(4;15)(q21;q15), and 46,XY,-6,-8,del(4)(q25q31),del(20)(q11.2) with additional der (8)(p22) (1 case) | SMA (+) in myofibroblasts; CD68 (+) in histiocytes | |
| Superficial acral fibromyxoma | RB1 deletions | Loss of RB1 expression; CD34 (+); CD99 (+); nestin (+) | |
| Cutaneous myxoma (superficial angiomyxoma) | PRKAR1A mutations (45%−65% of Carney complex); MYH8 mutations | Loss of PRKAR1A expression; SMA (+) | |
| Nodular fasciitis | USP6 rearrangements (~90%); MYH9::USP6 (>65%); PPP6R3::USP6 and CALD1::USP6 (malignant cases); EIF5A::USP6 (dermal nodular fasciitis with atypical mitosis) | SMA (+) | |
| Dermatofibrosarcoma protuberans | COL1A1::PDGFB (>90%); PDGFD rearrangements with COL6A3, EMILIN2, or TNC (<5%) | CD34 (+) | |
| Myxoinflammatory fibroblastic sarcoma | VGLL3 amplification (>50%); TGFBR3::MGEA5 (13%); BRAF fusions or amplifications | Cyclin D1 (+); factor XIIIa (+); podoplanin (D2-40) (+); CD10 (+); CD34 (±); CD163 (±); PRAME (±); CKs (±); CD68 (±); SMA (±) | |
| Myxofibrosarcoma | Frequent 5p gains with coamplification of TRIO, RICTOR, SKP2, and AMACR (associated with higher grade); inactivating mutations in TP53 (46%), RB1 (18%), and/or CDKN2A/2B (16%) | SMA (±); CD34 (±) | |
| Vascular tumors | Cherry hemangioma | Frequent somatic mutations of GNA14, GNA11, and GNAQ; rare somatic mutations in HRAS and KRAS | - |
| Spindle cell hemangioma | IDH1 p.R132C hotspot mutations (sporadic lesions and Maffucci syndrome); frequent somatic alterations in 2p22.3, 2q24.3, and 14q11.2 (Maffucci syndrome) | Endothelial markers (CD31, CD34, ERG) (+) in endothelial cells; SMA (±) and/or desmin (±) in spindle cells | |
| Epithelioid hemangioma | FOS or FOSB gene rearrangements | Endothelial markers (CD31, CD34, ERG) (+) in endothelial cells; FOSB (+) (FOSB-rearranged cases); SMA-positive pericytes | |
| Infantile hemangioma | No identified genetic abnormalities | GLUT1 (+) | |
| Congenital non-progressive hemangioma: RICH and NICH | Somatic activating mutations in GNAQ and GNA11 (RICH and NICH) | GLUT1 (–) | |
| Lobular capillary hemangioma | RAS-family and BRAF mutations (mainly c.1799T>A p.V600E) | - | |
| Verrucous venous malformation | Somatic missense mutation in MAP3K3 (some cases) | GLUT1 (±)a | |
| Arteriovenous malformation | Germline RASA1 mutations (hereditary CM-AVM syndrome) | GLUT1 (–) | |
| Lymphangioma (superficial lymphatic malformation) | Mutations in FLT4 (families with Milroy disease); mutations in PROX1 and FOXC2 | Lymphatic endothelial markers (podoplanin [D2-40], LYVE1, PROX1) (+); endothelial markers (CD31, ERG) (+) | |
| Postradiation atypical vascular lesion | Mutations in HRAS and TERT (rare cases) | Endothelial markers (CD31, CD34, ERG) (+); lymphatic endothelial markers (podoplanin [D2-40], LYVE1, PROX1) (+) | |
| Pseudomyogenic hemangioendothelioma | FOSB rearrangement, including SERPINE1::FOSB, ACTB::FOSB, WWTR1::FOSB, CLTC::FOSB, and EGFL7::FOSB (more aggressive) | CKs (+); FLI1 (+); ERG (+); FOSB (+); CD31 (+) (~50%); SMA (+) (~30%); CD34 (–) | |
| Epithelioid hemangioendothelioma | WWTR1::CAMTA1 (>90%); YAP1::TFE3 (rare subtype) | CAMTA1 (+) in EHE with WWTR1::CAMTA1 fusion; TFE3 (+) and loss of YAP1 C-terminus expression in EHE with YAP1::TFE3 fusion | |
| Composite hemangioendothelioma | YAP1::MAML2 (~27%); PTBP1::MAML2 (3 cases of neuroendocrine CHE); EPC1::PCH2 (1 case of neuroendocrine CHE) | Loss of YAP1 C-terminus expression in CHE with YAP1::MAML2 fusions; synaptophysin (+) in neuroendocrine CHE | |
| Kaposi sarcoma | Gain at 11q13 with FGF3 and FGF4 amplification; Y chromosome loss (early lesions); copy-number changes involving chromosomes 16, 17, 21, X, and Y (late lesions) | KSHV/HHV8-associated protein LANA-1 (+); CD31 (+); CD34 (+); ERG (+); podoplanin (D2-40) (+) | |
| Cutaneous angiosarcoma | Mutations in KDR, PTPRB, and PLCG1 (~40%); MYC amplification in >90% of radiation- and lymphedema-associated angiosarcoma;CIC alterations (younger patients) | MYC (+)b and frequent loss of H3K27me3 expression in radiation-induced angiosarcoma; CKs (+) and EMA (+) in epithelioid angiosarcoma | |
| Pericytic and perivascular tumors | Glomus tumor | Mutations in GLMN (familial); MIR143 and NOTCH genes (NOTCH1, NOTCH2, or NOTCH3) rearrangements (sporadic); BRAF p.V600E mutations (rare malignant tumors) | SMA (+); caldesmon (+); collagen IV (+); RGS5 (+) |
| Myopericytoma | BRAF mutations (15%) | SMA (+); caldesmon (+); MSA (+) | |
| Angioleiomyoma | Monosomy of chromosome 13; loss of 6p, 21q, and 13q; NOTCH gene fusions (very small subset) | SMA (+); calponin (+); caldesmon (+); desmin (±); RGS5 (+) | |
| Smooth muscle tumors | Cutaneous leiomyomas | Germline mutations in FH (HLRCC-associated tumors) | SMA (+); desmin (+); caldesmon (+); FH (–) and 2SC (+) in HLRCC-associated tumors |
| Neural tumors | Perineurioma | Deletion of 22q12, monosomies, and mutations in NF2; deletion of 17q11 (soft tissue subtype); 10q24 rearrangement (sclerosing subtype); loss of 13q and small deletions of chromosomes 3, 6, and 9 (malignant perineurioma) | EMA (+); collagen IV (+); laminin (+); claudin-1 (+); GLUT1 (+); CD34 (+) (~60%) |
| Neurofibroma | Mutations in NF1 gene (multiple or plexiform and diffuse subtypes); loss of CDKN2A (ANNUBP) | S100 protein (+); SOX 10 (+); CD34 (+) in perineurial fibroblasts; peripherin (+) in thin axons | |
| Schwannoma | NF2 mutations with complete or partial loss of chromosome 22; alterations in ARID1A/ARID1B (~29%), TSC1/TSC2 (~15%), or SH3PXD2A::HTRA1 (~10%); germline mutations in NF2, LZTR1, SMARCB1, and DGCR8 (associated with schwannoma susceptibility) | S100 protein (+); SOX10 (+); EMA (+) in perineurial capsule | |
| Granular cell tumor | Loss-of-function mutations in ATP6AP1 and ATP6AP2 (~72%) | S100 protein (+); SOX10 (+); CD68 (+); inhibin (±); calretinin (±); TFE3 (±); MITF (±) | |
| Malignant peripheral nerve sheath tumor | Mutations in NF1, CDKN2A/CDKN2B, and PRC2 core components (EED or SUZ12); complete loss of PRC2 activity and H3K27me3 expression (~80% of high-grade MPNSTs); SMARCB1 inactivation (~75% of epithelioid subtype) | S100 protein (+) (<50%); SOX10 (+) (<70%); GFAP (+) (<20%–30%); loss of H3K27me3 expressionc | |
| Tumors of uncertain differentiation | Non-neural granular cell tumor | ALK rearrangements (60%) | CD68 (+); CD63 (NKI/C3) (+); ALK (+) (subset) |
| Atypical fibroxanthoma | UV-radiation signature mutations in TP53; 9p and 13q deletions; TERT mutations | CD10 (+); CD68 (±); CD163 (±) | |
| Pleomorphic dermal sarcoma | Mutations in TP53, NOTCH family genes, and TERT promoter; alterations in CDKN2A and CDKN2B | CD10 (+); CD99 (+); PDGFRB (±); SMA (±); CD31 (±) | |
| Epithelioid sarcoma | Monoallelic or biallelic SMARCB1 deletion | Loss of SMARCB1 (INI1); CKs (+); EMA (+); CD34 (+) (>50%); ERG (+) (40%−67%) | |
| Dermal clear cell sarcoma | EWSR1::ATF1 (70%−90%); EWSR1::CREB1 (~5%) | S100 protein (+); SOX10 (+); PMEL (gp100) (+); TYRP1 (gp75) (+); melan-A (MART1) (+); MITF (+); PRAME (±)d | |
| Ewing sarcoma | EWSR1::FLI1 (~85%); EWSR1::ERG (5%−10%); EWSR1::FEV (rare); EWSR1::ETV1 (rare); FUS::ERG (rare) | CD99 (+); NKX2.2 (+); FLI1 (+) and ERG (+)e |
HMGA2, high mobility group AT-hook 2; RB1, retinoblastoma 1; MDM2, mouse double minute 2; CDK4, cyclin-dependent kinase 4; EGF, epidermal growth factor; PTEN, phosphatase and tensin homolog; FAP, familial adenomatous polyposis; FOSL1, FOS-like antigen 1; SMA, smooth muscle actin; PRKAR1A, protein kinase cAMP-dependent regulatory subunit type I alpha; PRAME, preferentially expressed antigen in melanoma; CK, cytokeratin; ERG, ETS-related gene; FOSB, FBJ murine osteosarcoma viral oncogene homolog B; GLUT1, glucose transporter 1; RICH, rapidly involuting congenital hemangioma; NICH, non-involuting congenital hemangioma; CM-AVM, capillary malformation–arteriovenous malformation; LYVE1, lymphatic vessel endothelial hyaluronan receptor 1; PROX1, prospero homeobox 1; FLI1, Friend leukemia virus integration 1; CAMTA1, calmodulin-binding transcription activator 1; EHE, epithelioid hemangioendothelioma; TFE3, transcription factor E3; YAP1 C-terminus, yes-associated protein 1 C-terminus; CHE, composite hemangioendothelioma; KSHV, Kaposi sarcoma-associated herpesvirus; HHV8, human herpesvirus 8; LANA-1, latency-associated nuclear antigen-1; MYC, MYC proto-oncogene; H3K27me3, histone H3 lysine 27 trimethylation; EMA, epithelial membrane antigen; RGS5, regulator of G-protein signaling 5; MSA, muscle-specific actin; HLRCC, hereditary leiomyomatosis and renal cell carcinoma; FH, fumarate hydratase; 2SC, 2-succinylcysteine; ANNUBP, atypical neurofibromatous neoplasm of uncertain biological potential; SOX10, SRY-box transcription factor 10; MITF, microphthalmia-associated transcription factor; PRC2, polycomb repressive complex 2; GFAP, glial fibrillary acidic protein; ALK, anaplastic lymphoma kinase; NOTCH, neurogenic locus notch homolog; PDGFRB, platelet-derived growth factor receptor beta; SMARCB1, SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily B, member 1; INI1, integrase interactor 1; PMEL, premelanosome protein; TYRP1, tyrosinase-related protein 1; MART1, melanoma antigen recognized by T cells 1; NKX2.2, NK2 homeobox 2; +, positive staining; ±, variable staining; –, negative staining.
aFocal and weak endothelial GLUT1 positivity may be seen;
bRadiation- and lymphedema-associated angiosarcomas are almost always positive for MYC but may occasionally be negative;
cComplete loss of H3K27me3 expression occurs more frequently in high-grade and radiation-induced MPNSTs than in low-grade MPNSTs;
dPRAME expression is usually absent or only focal;
eFLI1 and ERG are often expressed in cases with the corresponding translocation.
- 1. WHO Classification of Tumours Editorial Board. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025.
- 2. Elder DE, Massi D, Scolyer RA, Willemze R. WHO classification of skin tumours. 4th ed. Lyon: IARC Press, 2018.
- 3. Papke DJ Jr, Hornick JL. Recent advances in the diagnosis, classification and molecular pathogenesis of cutaneous mesenchymal neoplasms. Histopathology 2022; 80: 216-32. ArticlePubMedPDF
- 4. Fischer GM, Papke DJ. Gene fusions in superficial mesenchymal neoplasms: emerging entities and useful diagnostic adjuncts. Semin Diagn Pathol 2023; 40: 246-57. ArticlePubMed
- 5. Kalmykova AV, Baranovska-Andrigo V, Michal M. Update on cutaneous mesenchymal tumors in the 5th edition of WHO classification of skin tumors with an emphasis on new fusion-associated neoplasms. Virchows Arch 2024; 485: 777-92. ArticlePubMedPMCPDF
- 6. Ho J, Collie CJ. What's new in dermatopathology 2023: WHO 5th edition updates. J Pathol Transl Med 2023; 57: 337-40. ArticlePubMedPMCPDF
- 7. Requena L, Cheah AL, Patel RM. Naevus lipomatosus superficialis. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 459-60.
- 8. Low I, Fritchie KJ, Ivan D. Lipoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 461-2.
- 9. Low I, Calonje JE, Ivan D. Lipoma. In: Elder DE, Massi D, Scolyer RA, Willemze R, eds. WHO classification of skin tumours. 4th ed. Lyon: IARC Press, 2018; 296-7.
- 10. Billings SD, Requena L. Angiolipoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 463-4.
- 11. Sheng W, Lu L, Wang J. Cellular angiolipoma: a clinicopathological and immunohistochemical study of 12 cases. Am J Dermatopathol 2013; 35: 220-5. ArticlePubMed
- 12. Patel RM, Billings SD, Ud Din N, Wong D. Spindle cell/pleomorphic lipoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 465-6.
- 13. Folpe AL, Gill AJ. Atypical lipomatous tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 467-8.
- 14. Mathew R, Morgan MB. Dermal atypical lipomatous tumor/well-differentiated liposarcoma obfuscated by epidermal inclusion cyst: a wolf in sheep's clothing? Am J Dermatopathol 2006; 28: 338-40. ArticlePubMed
- 15. Paredes BE, Mentzel T. Atypical lipomatous tumor/"well-differentiated liposarcoma" of the skin clinically presenting as a skin tag: clinicopathologic, immunohistochemical, and molecular analysis of 2 cases. Am J Dermatopathol 2011; 33: 603-7. ArticlePubMed
- 16. Yoshida A, Ushiku T, Motoi T, Shibata T, Fukayama M, Tsuda H. Well-differentiated liposarcoma with low-grade osteosarcomatous component: an underrecognized variant. Am J Surg Pathol 2010; 34: 1361-6. ArticlePubMed
- 17. Evans HL. Atypical lipomatous tumor, its variants, and its combined forms: a study of 61 cases, with a minimum follow-up of 10 years. Am J Surg Pathol 2007; 31: 1-14. ArticlePubMed
- 18. Patel RM, Folpe AL. Pleomorphic liposarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 469-70.
- 19. Miettinen M, Enzinger FM. Epithelioid variant of pleomorphic liposarcoma: a study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod Pathol 1999; 12: 722-8. PubMed
- 20. Pizem J, Matjasic A, Zupan A, Luzar B, Sekoranja D, Dimnik K. Fibroma of tendon sheath is defined by a USP6 gene fusion-morphologic and molecular reappraisal of the entity. Mod Pathol 2021; 34: 1876-88. ArticlePubMedPDF
- 21. Eisenberg JM, Buckwalter V JA, Snow AN, Davick J. Cellular fibroma of tendon sheath with novel TNC-USP6 gene fusion clinically mimicking arthritis in a 7-year-old boy. Pediatr Dev Pathol 2022; 25: 192-6. ArticlePubMedPDF
- 22. Mantilla JG, Gross JM, Liu YJ, Hoch BL, Ricciotti RW. Characterization of novel USP6 gene rearrangements in a subset of so-called cellular fibroma of tendon sheath. Mod Pathol 2021; 34: 13-9. ArticlePubMedPDF
- 23. Puls F, Hofvander J, Magnusson L, et al. Fn1-EGF gene fusions are recurrent in calcifying aponeurotic fibroma. J Pathol 2016; 238: 502-7. ArticlePubMed
- 24. Al-Ibraheemi A, Folpe AL, Perez-Atayde AR, et al. Aberrant receptor tyrosine kinase signaling in lipofibromatosis: a clinicopathological and molecular genetic study of 20 cases. Mod Pathol 2019; 32: 423-34. ArticlePubMedPDF
- 25. Fetsch JF, Miettinen M. Calcifying aponeurotic fibroma: a clinicopathologic study of 22 cases arising in uncommon sites. Hum Pathol 1998; 29: 1504-10. ArticlePubMed
- 26. Wang WL, Al-Zaid T, Patel RM. Sclerotic fibroma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 475.
- 27. Cloutier JM, Shalin SC, Lindberg M, et al. Cutaneous pleomorphic fibromas arising in patients with germline TP53 mutations. J Cutan Pathol 2020; 47: 734-41. ArticlePDF
- 28. Linos K, Luzar B, O'Brien BH. Dermatofibroma (fibrous histiocytoma). In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 488-90.
- 29. Plaszczyca A, Nilsson J, Magnusson L, et al. Fusions involving protein kinase C and membrane-associated proteins in benign fibrous histiocytoma. Int J Biochem Cell Biol 2014; 53: 475-81. ArticlePubMed
- 30. Jedrych JJ, Duraisamy S, Karunamurthy A. Aneurysmal fibrous histiocytomas with recurrent rearrangement of the PRKCD gene and LAMTOR1-PRKCD fusions. J Cutan Pathol 2018; 45: 966-8. ArticlePubMedPDF
- 31. Panagopoulos I, Gorunova L, Bjerkehagen B, Lobmaier I, Heim S. LAMTOR1-PRKCD and NUMA1-SFMBT1 fusion genes identified by RNA sequencing in aneurysmal benign fibrous histiocytoma with t(3;11)(p21;q13). Cancer Genet 2015; 208: 545-51. ArticlePubMed
- 32. Prieto VG, Andea AA, Nascimento AF. Superficial fibromatosis. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 491-2.
- 33. Montgomery E, Lee JH, Abraham SC, Wu TT. Superficial fibromatoses are genetically distinct from deep fibromatoses. Mod Pathol 2001; 14: 695-701. ArticlePubMedPDF
- 34. Redlich GC, Montgomery KD, Allgood GA, Joste NE. Plexiform fibrohistiocytic tumor with a clonal cytogenetic anomaly. Cancer Genet Cytogenet 1999; 108: 141-3. ArticlePubMed
- 35. Smith S, Fletcher CD, Smith MA, Gusterson BA. Cytogenetic analysis of a plexiform fibrohistiocytic tumor. Cancer Genet Cytogenet 1990; 48: 31-4. ArticlePubMed
- 36. Leclerc-Mercier S, Pedeutour F, Fabas T, Glorion C, Brousse N, Fraitag S. Plexiform fibrohistiocytic tumor with molecular and cytogenetic analysis. Pediatr Dermatol 2011; 28: 26-9. ArticlePubMed
- 37. Requena L. Cutaneous myxoma (superficial angiomyxoma). In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 499-500.
- 38. Veugelers M, Wilkes D, Burton K, et al. Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and prkar1a haploinsufficient mice. Proc Natl Acad Sci U S A 2004; 101: 14222-7. ArticlePubMedPMC
- 39. Wilkes D, McDermott DA, Basson CT. Clinical phenotypes and molecular genetic mechanisms of Carney complex. Lancet Oncol 2005; 6: 501-8. ArticlePubMed
- 40. Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest 2011; 91: 1427-33. ArticlePubMedPDF
- 41. Teramura Y, Yamazaki Y, Tanaka M, et al. Case of mesenchymal tumor with the PPP6R3-USP6 fusion, possible nodular fasciitis with malignant transformation. Pathol Int 2019; 69: 706-9. ArticlePubMedPDF
- 42. Papke DJ Jr, Oliveira AM, Chou MM, Fletcher CD. Morphologically malignant nodular fasciitis with CALD1-USP6 fusion. Virchows Arch 2021; 479: 1007-12. ArticlePubMedPDF
- 43. Lenz J, Michal M, Svajdler M, et al. Novel EIF5A-USP6 gene fusion in nodular fasciitis associated with unusual pathologic features: a report of a case and review of the literature. Am J Dermatopathol 2020; 42: 539-43. ArticlePubMed
- 44. Dadone-Montaudie B, Alberti L, Duc A, et al. Alternative PDGFD rearrangements in dermatofibrosarcomas protuberans without PDGFB fusions. Mod Pathol 2018; 31: 1683-93. ArticlePubMedPDF
- 45. Lee PH, Huang SC, Wu PS, et al. Molecular characterization of dermatofibrosarcoma protuberans: the clinicopathologic significance of uncommon fusion gene rearrangements and their diagnostic importance in the exclusively subcutaneous and circumscribed lesions. Am J Surg Pathol 2022; 46: 942-55. ArticlePubMed
- 46. Abbott JJ, Erickson-Johnson M, Wang X, Nascimento AG, Oliveira AM. Gains of COL1A1-PDGFB genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Mod Pathol 2006; 19: 1512-8. ArticlePubMedPDF
- 47. Hiraki-Hotokebuchi Y, Yamada Y, Kohashi K, et al. Alteration of PDGFRβ-Akt-mTOR pathway signaling in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Hum Pathol 2017; 67: 60-8. ArticlePubMed
- 48. Wang WL, Colebatch AJ, Gill AJ, Wang J. Dermatofibrosarcoma protuberans. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 511-4.
- 49. Arbajian E, Hofvander J, Magnusson L, Mertens F. Deep sequencing of myxoinflammatory fibroblastic sarcoma. Genes Chromosomes Cancer 2020; 59: 309-17. ArticlePubMedPDF
- 50. Suster D, Michal M, Huang H, et al. Myxoinflammatory fibroblastic sarcoma: an immunohistochemical and molecular genetic study of 73 cases. Mod Pathol 2020; 33: 2520-33. ArticlePubMedPDF
- 51. Antonescu CR, Zhang L, Nielsen GP, Rosenberg AE, Dal Cin P, Fletcher CD. Consistent t(1;10) with rearrangements of TGFBR3 and MGEA5 in both myxoinflammatory fibroblastic sarcoma and hemosiderotic fibrolipomatous tumor. Genes Chromosomes Cancer 2011; 50: 757-64. ArticlePubMedPMC
- 52. Zreik RT, Carter JM, Sukov WR, et al. TGFBR3 and MGEA5 rearrangements are much more common in "hybrid" hemosiderotic fibrolipomatous tumor-myxoinflammatory fibroblastic sarcomas than in classical myxoinflammatory fibroblastic sarcomas: a morphological and fluorescence in situ hybridization study. Hum Pathol 2016; 53: 14-24. ArticlePubMed
- 53. Carter JM, Sukov WR, Montgomery E, et al. TGFBR3 and MGEA5 rearrangements in pleomorphic hyalinizing angiectatic tumors and the spectrum of related neoplasms. Am J Surg Pathol 2014; 38: 1182-992. ArticlePubMed
- 54. Kao YC, Ranucci V, Zhang L, et al. Recurrent BRAF gene rearrangements in myxoinflammatory fibroblastic sarcomas, but not hemosiderotic fibrolipomatous tumors. Am J Surg Pathol 2017; 41: 1456-65. ArticlePubMedPMC
- 55. Huang HY, Billings SD, Creytens D, Shibata T. Myxofibrosarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 517-8.
- 56. Willems SM, Debiec-Rychter M, Szuhai K, Hogendoorn PC, Sciot R. Local recurrence of myxofibrosarcoma is associated with increase in tumour grade and cytogenetic aberrations, suggesting a multistep tumour progression model. Mod Pathol 2006; 19: 407-16. ArticlePubMedPDF
- 57. Cancer Genome Atlas Research Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017; 171: 950-65. ArticlePubMedPMC
- 58. Ogura K, Hosoda F, Arai Y, et al. Integrated genetic and epigenetic analysis of myxofibrosarcoma. Nat Commun 2018; 9: 2765.ArticlePubMedPMCPDF
- 59. Li CF, Wang JM, Kang HY, et al. Characterization of gene amplification-driven SKP2 overexpression in myxofibrosarcoma: potential implications in tumor progression and therapeutics. Clin Cancer Res 2012; 18: 1598-610. ArticlePubMedPDF
- 60. Li CF, Fang FM, Lan J, et al. AMACR amplification in myxofibrosarcomas: a mechanism of overexpression that promotes cell proliferation with therapeutic relevance. Clin Cancer Res 2014; 20: 6141-52. ArticlePubMedPDF
- 61. Okada T, Lee AY, Qin LX, et al. Integrin-α10 dependency identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discov 2016; 6: 1148-65. ArticlePubMedPMCPDF
- 62. Calonje JE, Panse G. Fibrous papule. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 483-4.
- 63. Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol 2018; 40: 551-60. ArticlePubMed
- 64. Schaffer JV, Gohara MA, McNiff JM, Aasi SZ, Dvoretzky I. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol 2005; 53(2 Suppl 1): S108-11. ArticlePubMed
- 65. Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 1997; 133: 853-7. ArticlePubMed
- 66. Chan JY, Wang KH, Fang CL, Chen WY. Fibrous papule of the face, similar to tuberous sclerosis complex-associated angiofibroma, shows activation of the mammalian target of rapamycin pathway: evidence for a novel therapeutic strategy? PLoS One 2014; 9: e89467. ArticlePubMedPMC
- 67. Bansal C, Stewart D, Li A, Cockerell CJ. Histologic variants of fibrous papule. J Cutan Pathol 2005; 32: 424-8. ArticlePubMed
- 68. Krauland K, Patel AB. Junctional melanocytic hyperplasia overlying benign fibrous papule: an under-recognized variant of a common lesion. J Cutan Pathol 2020; 47: 1220-3. ArticlePubMedPDF
- 69. Fraitag Spinner S, Billings SD. Fibroblastic connective tissue naevus. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 485-6.
- 70. de Feraudy S, Fletcher CD. Fibroblastic connective tissue nevus: a rare cutaneous lesion analyzed in a series of 25 cases. Am J Surg Pathol 2012; 36: 1509-15. ArticlePubMed
- 71. Pennacchia I, Kutzner H, Kazakov DV, Mentzel T. Fibroblastic connective tissue nevus: clinicopathological and immunohistochemical study of 14 cases. J Cutan Pathol 2017; 44: 827-34. ArticlePubMedPDF
- 72. Velez MJ, Billings SD, Weaver JA. Fibroblastic connective tissue nevus. J Cutan Pathol 2016; 43: 75-9. ArticlePubMed
- 73. Tallegas M, Fraitag S, Binet A, et al. Novel KHDRBS1-NTRK3 rearrangement in a congenital pediatric CD34-positive skin tumor: a case report. Virchows Arch 2019; 474: 111-5. ArticlePubMedPDF
- 74. Lynch MC, Samson TD, Zaenglein AL, Chung CG. Evolution of fibroblastic connective tissue nevus in an infant. Am J Dermatopathol 2017; 39: 225-7. ArticlePubMed
- 75. Ohata M, Fujiwara S, Yoshioka A, et al. Agminated fibroblastic connective tissue nevus in a 1-year-old boy. Pediatr Dermatol 2019; 36: 997-8. ArticlePubMedPDF
- 76. Downey C, Requena L, Bague S, Sanchez Martinez MA, Lloreta J, Baselga E. Agminated fibroblastic connective tissue nevus: a new clinical presentation. Pediatr Dermatol 2016; 33: e240-3. ArticlePubMed
- 77. Allemant Ortiz LJ, Calderon-Castrat X, Orellana Cortez A, Ingar Carbone B, Santos-Briz A. Congenital atrophic plaque: fibroblastic connective tissue nevus. Pediatr Dermatol 2017; 34: e216-8. ArticlePubMed
- 78. Oliveira AM, Rosenberg AE. Fibro-osseous tumour of digits. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 487.
- 79. Dupree WB, Enzinger FM. Fibro-osseous pseudotumor of the digits. Cancer 1986; 58: 2103-9. ArticlePubMed
- 80. Flucke U, Shepard SJ, Bekers EM, et al. Fibro-osseous pseudotumor of digits: expanding the spectrum of clonal transient neoplasms harboring USP6 rearrangement. Ann Diagn Pathol 2018; 35: 53-5. ArticlePubMed
- 81. Svajdler M, Michal M, Martinek P, et al. Fibro-osseous pseudotumor of digits and myositis ossificans show consistent COL1A1-USP6 rearrangement: a clinicopathological and genetic study of 27 cases. Hum Pathol 2019; 88: 39-47. ArticlePubMed
- 82. de Silva MV, Reid R. Myositis ossificans and fibroosseous pseudotumor of digits: a clinicopathological review of 64 cases with emphasis on diagnostic pitfalls. Int J Surg Pathol 2003; 11: 187-95. ArticlePubMedPDF
- 83. Wang JC, Li WS, Kao YC, et al. Clinicopathological and molecular characterisation of USP6-rearranged soft tissue neoplasms: the evidence of genetic relatedness indicates an expanding family with variable bone-forming capacity. Histopathology 2021; 78: 676-89. ArticlePubMedPDF
- 84. Rosenberg AE. Pseudosarcomas of soft tissue. Arch Pathol Lab Med 2008; 132: 579-86. ArticlePubMedPDF
- 85. Laskin WB. Inclusion body fibromatosis. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 493-4.
- 86. Coffin CM, Dehner LP. Fibroblastic-myofibroblastic tumors in children and adolescents: a clinicopathologic study of 108 examples in 103 patients. Pediatr Pathol 1991; 11: 569-88. ArticlePubMed
- 87. Chung EB. Pitfalls in diagnosing benign soft tissue tumors in infancy and childhood. Pathol Annu 1985; 20 Pt 2: 323-86. PubMed
- 88. Plusje LG, Bastiaens M, Chang A, Hogendoorn PC. Infantile-type digital fibromatosis tumour in an adult. Br J Dermatol 2000; 143: 1107-8. ArticlePubMedPDF
- 89. Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma/fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol 2009; 33: 1-13. ArticlePubMed
- 90. Purdy LJ, Colby TV. Infantile digital fibromatosis occurring outside the digit. Am J Surg Pathol 1984; 8: 787-90. ArticlePubMed
- 91. Allen PW. Recurring digital fibrous tumours of childhood. Pathology 1972; 4: 215-23. ArticlePubMed
- 92. Mukai M, Torikata C, Iri H, Hata J, Naito M, Shimoda T. Immunohistochemical identification of aggregated actin filaments in formalin-fixed, paraffin-embedded sections. I. A study of infantile digital fibromatosis by a new pretreatment. Am J Surg Pathol 1992; 16: 110-5. ArticlePubMed
- 93. Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology 2014; 64: 752-5. ArticlePubMed
- 94. McNiff J, Roy SF. Multinucleate cell angiohistiocytoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 503-4.
- 95. Cribier B, Gambini C, Rainero M, Grosshans E. Multinucleate cell angiohistiocytoma: a review and report of four cases. Acta Derm Venereol 1995; 75: 337-9. ArticlePubMedPDF
- 96. Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol 2015; 37: 222-8. ArticlePubMed
- 97. Roy SF, Dong D, Myung P, McNiff JM. Multinucleate cell angiohistiocytoma: a clinicopathologic study of 62 cases and proposed diagnostic criteria. J Cutan Pathol 2019; 46: 563-9. ArticlePubMedPDF
- 98. Jia QN, Qiao J, Qu T. Generalized multinucleate cell angiohistiocytoma with possible origin from fibroblasts: a clinicopathological study of 15 cases. J Dermatol 2021; 48: 114-9. ArticlePubMedPDF
- 99. Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol 2010; 32: 655-9. ArticlePubMed
- 100. Jaconelli L, Kanitakis J, Ktiouet S, Faure M, Claudy A. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J 2009; 15: 4.Article
- 101. Suurmeijer AJ, Habeeb O, Michael M, Thway K. EWSR1::SMAD3-rearranged fibroblastic tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 509-10.
- 102. Michal M, Berry RS, Rubin BP, et al. EWSR1-SMAD3-rearranged fibroblastic tumor: an emerging entity in an increasingly more complex group of fibroblastic/myofibroblastic neoplasms. Am J Surg Pathol 2018; 42: 1325-33. ArticlePubMed
- 103. Habeeb O, Korty KE, Azzato EM, et al. EWSR1-SMAD3 rearranged fibroblastic tumor: case series and review. J Cutan Pathol 2021; 48: 255-62. ArticlePubMedPDF
- 104. Calonje JE, Marusic Z, North PE, Sangueza OP. Tufted haemangioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 530-1.
- 105. Ten Broek RW, Koelsche C, Eijkelenboom A, et al. Kaposiform hemangioendothelioma and tufted angioma: (epi)genetic analysis including genome-wide methylation profiling. Ann Diagn Pathol 2020; 44: 151434.ArticlePubMed
- 106. Requena L, Calonje JE, Fisher C. Postradiation atypical vascular lesion. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 551-2.
- 107. Hornick JL, Billings SD, Calonje JE, Folpe AL, Requena L. Hobnail haemangioendotheliomas. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 557-8.
- 108. Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J Invest Dermatol 2016; 136: 481-6. ArticlePubMed
- 109. Jansen P, Muller H, Lodde GC, et al. GNA14, GNA11, and GNAQ mutations are frequent in benign but not malignant cutaneous vascular tumors. Front Genet 2021; 12: 663272.ArticlePubMedPMC
- 110. Liau JY, Lee JC, Tsai JH, Chen CC, Chung YC, Wang YH. High frequency of GNA14, GNAQ, and GNA11 mutations in cherry hemangioma: a histopathological and molecular study of 85 cases indicating GNA14 as the most commonly mutated gene in vascular neoplasms. Mod Pathol 2019; 32: 1657-65. ArticlePubMedPDF
- 111. Legendre P, Norkowski E, Le Pelletier F, et al. Glomeruloid haemangioma: a possible consequence of elevated VEGF in POEMS and Erdheim-Chester disease. Eur J Dermatol 2018; 28: 784-9. ArticlePubMedPDF
- 112. Calonje JE, Marusic Z, North PE, Sangueza OP. Epithelioid haemangioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 528-9.
- 113. Ortins-Pina A, Llamas-Velasco M, Turpin S, Soares-de-Almeida L, Filipe P, Kutzner H. FOSB immunoreactivity in endothelia of epithelioid hemangioma (angiolymphoid hyperplasia with eosinophilia). J Cutan Pathol 2018; 45: 395-402. ArticlePubMedPDF
- 114. Calonje JE, Marusic Z, North PE, Sangueza OP. Arteriovenous malformation. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 545-6.
- 115. Billings SD, Agaram NP, Calonje JE, Folpe AL, Hornick JL, Requena L. Pseudomyogenic haemangioendothelioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 553-4.
- 116. Walther C, Tayebwa J, Lilljebjorn H, et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J Pathol 2014; 232: 534-40. ArticlePubMed
- 117. Zhu G, Benayed R, Ho C, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol 2019; 32: 609-20. ArticlePubMedPDF
- 118. Panagopoulos I, Lobmaier I, Gorunova L, Heim S. Fusion of the genes WWTR1 and FOSB in pseudomyogenic hemangioendothelioma. Cancer Genomics Proteomics 2019; 16: 293-8. ArticlePubMedPMC
- 119. Bridge JA, Sumegi J, Royce T, Baker M, Linos K. A novel CLTC-FOSB gene fusion in pseudomyogenic hemangioendothelioma of bone. Genes Chromosomes Cancer 2021; 60: 38-42. ArticlePubMed
- 120. Hakar MH, White K, Hansford BG, Swensen J, Davis JL. Novel EGFL7-FOSB fusion in pseudomyogenic haemangioendothelioma with widely metastatic disease. Histopathology 2021; 79: 888-91. ArticlePubMedPDF
- 121. Hornick JL, Billings SD, Calonje JE, Folpe AL, Requena L. Epithelioid haemangioendothelioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 555-6.
- 122. Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011; 50: 644-53. ArticlePubMedPMC
- 123. Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013; 52: 775-84. ArticlePubMedPMC
- 124. Dermawan JK, Azzato EM, Billings SD, et al. YAP1-TFE3-fused hemangioendothelioma: a multi-institutional clinicopathologic study of 24 genetically-confirmed cases. Mod Pathol 2021; 34: 2211-21. ArticlePubMedPDF
- 125. Anderson WJ, Fletcher CD, Hornick JL. Loss of expression of YAP1 C-terminus as an ancillary marker for epithelioid hemangioendothelioma variant with YAP1-TFE3 fusion and other YAP1-related vascular neoplasms. Mod Pathol 2021; 34: 2036-42. ArticlePubMedPDF
- 126. Hornick JL, Billings SD, Folpe AL, Requena L, Rubin BP. Composite haemangioendothelioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 559-60.
- 127. Antonescu CR, Dickson BC, Sung YS, et al. Recurrent YAP1 and MAML2 gene rearrangements in retiform and composite hemangioendothelioma. Am J Surg Pathol 2020; 44: 1677-84. ArticlePubMedPMC
- 128. Perry KD, Al-Lbraheemi A, Rubin BP, et al. Composite hemangioendothelioma with neuroendocrine marker expression: an aggressive variant. Mod Pathol 2017; 30: 1589-602. ArticlePubMedPDF
- 129. Dermawan JK, Westra WH, Antonescu CR. Recurrent PTBP1::MAML2 fusions in composite hemangioendothelioma with neuroendocrine differentiation: a report of two cases involving neck lymph nodes. Genes Chromosomes Cancer 2022; 61: 187-93. ArticlePubMed
- 130. Grayson W, Dezube BJ, Doyle LA, Pantanowitz L. Kaposi sarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 561-5.
- 131. Kiuru-Kuhlefelt S, Sarlomo-Rikala M, Larramendy ML, et al. FGF4 and INT2 oncogenes are amplified and expressed in Kaposi's sarcoma. Mod Pathol 2000; 13: 433-7. ArticlePubMedPDF
- 132. Pyakurel P, Montag U, Castanos-Velez E, et al. CGH of microdissected Kaposi's sarcoma lesions reveals recurrent loss of chromosome Y in early and additional chromosomal changes in late tumour stages. AIDS 2006; 20: 1805-12. ArticlePubMed
- 133. Billings SD, Brenn T, Hornick JL, Thway K. Cutaneous angiosarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 566-7.
- 134. Painter CA, Jain E, Tomson BN, et al. The angiosarcoma project: enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat Med 2020; 26: 181-7. ArticlePubMedPDF
- 135. Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol 2010; 176: 34-9. ArticlePubMedPMC
- 136. Ko JS, Billings SD, Lanigan CP, Buehler D, Fernandez AP, Tubbs RR. Fully automated dual-color dual-hapten silver in situ hybridization staining for MYC amplification: a diagnostic tool for discriminating secondary angiosarcoma. J Cutan Pathol 2014; 41: 286-92. ArticlePubMed
- 137. Fernandez AP, Sun Y, Tubbs RR, Goldblum JR, Billings SD. Fish for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol 2012; 39: 234-42. ArticlePubMed
- 138. Winderlich M, Keller L, Cagna G, et al. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol 2009; 185: 657-71. ArticlePubMedPMCPDF
- 139. Behjati S, Tarpey PS, Sheldon H, et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet 2014; 46: 376-9. ArticlePubMedPMC
- 140. Antonescu CR, Yoshida A, Guo T, et al. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res 2009; 69: 7175-9. ArticlePubMedPMCPDF
- 141. Torrence D, Antonescu CR. The genetics of vascular tumours: an update. Histopathology 2022; 80: 19-32. ArticlePubMedPMCPDF
- 142. Huang SC, Zhang L, Sung YS, et al. Recurrent CIC gene abnormalities in angiosarcomas: a molecular study of 120 cases with concurrent investigation of PLCG1, KDR, MYC, and FLT4 gene alterations. Am J Surg Pathol 2016; 40: 645-55. ArticlePubMedPMC
- 143. Calonje JE, Marusic Z, North PE, Sangueza OP. Papillary haemangioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 525.
- 144. Suurmeijer AJ, Fletcher CD. Papillary haemangioma: a distinctive cutaneous haemangioma of the head and neck area containing eosinophilic hyaline globules. Histopathology 2007; 51: 638-48. ArticlePubMed
- 145. Maloney N, Miller P, Linos K. Papillary hemangioma: an under-recognized entity not to be confused with glomeruloid hemangioma. Am J Dermatopathol 2020; 42: 211-4. ArticlePubMed
- 146. Marusic Z, Calonje JE, North PE, Sangueza OP. Poikilodermatous plaque-like haemangioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 540-1.
- 147. Semkova K, Carr R, Grainger M, et al. Poikilodermatous plaque-like hemangioma: case series of a newly defined entity. J Am Acad Dermatol 2019; 81: 1257-70. ArticlePubMed
- 148. Sullivan M, Hartman R, Mahalingam M. Poikilodermatous plaque-like hemangioma: a benign vasoformative entity with reproducible histopathologic and clinical features. J Cutan Pathol 2020; 47: 950-3. ArticlePubMedPDF
- 149. Calonje JE, Marusic Z, North PE, Sangueza OP. Acquired elastotic haemangioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 542.
- 150. Requena L, Kutzner H, Mentzel T. Acquired elastotic hemangioma: a clinicopathologic variant of hemangioma. J Am Acad Dermatol 2002; 47: 371-6. ArticlePubMed
- 151. Jeunon T, Carvalho Wagnes Stofler ME, Teixeira Rezende P, Staccioli Castro M, Jeunon-Sousa MA. Acquired elastotic hemangioma: a case report and review of 49 previously reported cases. Am J Dermatopathol 2020; 42: 244-50. ArticlePubMed
- 152. Fanburg-Smith JC, Michal M, Partanen TA, Alitalo K, Miettinen M. Papillary intralymphatic angioendothelioma (PILA): a report of twelve cases of a distinctive vascular tumor with phenotypic features of lymphatic vessels. Am J Surg Pathol 1999; 23: 1004-10. ArticlePubMed
- 153. Li B, Li Y, Tian XY, Li Z. Unusual multifocal intraosseous papillary intralymphatic angioendothelioma (Dabska tumor) of facial bones: a case report and review of literature. Diagn Pathol 2013; 8: 160.ArticlePubMedPMCPDF
- 154. Gambarotti M, Righi A, Sbaraglia M, et al. Intraosseous papillary intralymphatic angioendothelioma (PILA): one new case and review of the literature. Clin Sarcoma Res 2018; 8: 1.ArticlePubMedPMCPDF
- 155. Calonje E, Fletcher CD, Wilson-Jones E, Rosai J. Retiform hemangioendothelioma: a distinctive form of low-grade angiosarcoma delineated in a series of 15 cases. Am J Surg Pathol 1994; 18: 115-25. PubMed
- 156. Tan D, Kraybill W, Cheney RT, Khoury T. Retiform hemangioendothelioma: a case report and review of the literature. J Cutan Pathol 2005; 32: 634-7. ArticlePubMed
- 157. Arriola AG, Taylor LA, Asemota E, et al. Atypical retiform hemangioendothelioma arising in a patient with Milroy disease: a case report and review of the literature. J Cutan Pathol 2017; 44: 98-103. ArticlePubMedPDF
- 158. Emberger M, Laimer M, Steiner H, Zelger B. Retiform hemangioendothelioma: presentation of a case expressing D2-40. J Cutan Pathol 2009; 36: 987-90. ArticlePubMed
- 159. Miettinen M, Wang ZF. Prox1 transcription factor as a marker for vascular tumors-evaluation of 314 vascular endothelial and 1086 nonvascular tumors. Am J Surg Pathol 2012; 36: 351-9. ArticlePubMedPMC
- 160. Jackett LA, Folpe AL. Glomus tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 568-70.
- 161. Mosquera JM, Sboner A, Zhang L, et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer 2013; 52: 1075-87. ArticlePubMedPMC
- 162. Agaram NP, Zhang L, Jungbluth AA, Dickson BC, Antonescu CR. A molecular reappraisal of glomus tumors and related pericytic neoplasms with emphasis on NOTCH-gene fusions. Am J Surg Pathol 2020; 44: 1556-62. ArticlePubMedPMC
- 163. Chakrapani A, Warrick A, Nelson D, Beadling C, Corless CL. BRAF and KRAS mutations in sporadic glomus tumors. Am J Dermatopathol 2012; 34: 533-5. ArticlePubMed
- 164. Karamzadeh Dashti N, Bahrami A, Lee SJ, et al. BRAF V600E mutations occur in a subset of glomus tumors, and are associated with malignant histologic characteristics. Am J Surg Pathol 2017; 41: 1532-41. ArticlePubMed
- 165. Agaimy A, Agaram NP, Luzar B, Michal M. Myopericytoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 571-2.
- 166. Dahlen A, Fletcher CD, Mertens F, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12). Am J Pathol 2004; 164: 1645-53. ArticlePubMedPMC
- 167. Antonescu CR, Sung YS, Zhang L, Agaram NP, Fletcher CD. Recurrent SRF-RELA fusions define a novel subset of cellular myofibroma/myopericytoma: a potential diagnostic pitfall with sarcomas with myogenic differentiation. Am J Surg Pathol 2017; 41: 677-84. ArticlePubMedPMC
- 168. Patel RM, Agaram NP, Michal M, Requena L. Angioleiomyoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 575-6.
- 169. Welborn J, Fenner S, Parks R. Angioleiomyoma: a benign tumor with karyotypic aberrations. Cancer Genet Cytogenet 2010; 199: 147-8. ArticlePubMed
- 170. Shen J, Shrestha S, Yen YH, et al. The pericyte antigen RGS5 in perivascular soft tissue tumors. Hum Pathol 2016; 47: 121-31. ArticlePubMed
- 171. Agaimy A, Agaram NP, Luzar B, Michal M. Myofibroma and myofibromatosis. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 573-4.
- 172. Oudijk L, den Bakker MA, Hop WC, et al. Solitary, multifocal and generalized myofibromas: clinicopathological and immunohistochemical features of 114 cases. Histopathology 2012; 60: E1-11. ArticlePubMed
- 173. Martignetti JA, Tian L, Li D, et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet 2013; 92: 1001-7. ArticlePubMedPMC
- 174. Agaimy A, Bieg M, Michal M, et al. Recurrent somatic PDGFRB mutations in sporadic infantile/solitary adult myofibromas but not in angioleiomyomas and myopericytomas. Am J Surg Pathol 2017; 41: 195-203. ArticlePubMed
- 175. Nihous H, Macagno N, Baud-Massiere J, et al. Genetic variant of SRF-rearranged myofibroma with a misleading nuclear expression of STAT6 and STAT6 involvement as 3' fusion partner. Virchows Arch 2021; 478: 597-603. ArticlePubMedPDF
- 176. Folpe AL, Elston D. Atypical intradermal smooth muscle neoplasm. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 583-4.
- 177. Kraft S, Fletcher CD. Atypical intradermal smooth muscle neoplasms: clinicopathologic analysis of 84 cases and a reappraisal of cutaneous "leiomyosarcoma". Am J Surg Pathol 2011; 35: 599-607. ArticlePubMed
- 178. Spinner S, Davis JL. Smooth muscle hamartoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 577-8.
- 179. Gonzalez IA, Dehner LP. Smooth muscle hamartoma and striated muscle hamartoma: clinicopathologic characterization of two rare entities and literature review. J Cutan Pathol 2021; 48: 237-46. ArticlePubMedPDF
- 180. Garcia-Gavin J, Perez-Perez L, Allegue F, Perez-Pedrosa A, Ortiz-Rey JA, Zulaica A. Multiple congenital familial smooth muscle hamartoma in two siblings. Dermatol Online J 2012; 18: 7.
- 181. Schnur RE, Herzberg AJ, Spinner N, et al. Variability in the Michelin tire syndrome: a child with multiple anomalies, smooth muscle hamartoma, and familial paracentric inversion of chromosome 7q. J Am Acad Dermatol 1993; 28: 364-70. PubMed
- 182. Zarineh A, Kozovska ME, Brown WG, Elder DE, Rabkin MS. Smooth muscle hamartoma associated with a congenital pattern melanocytic nevus, a case report and review of the literature. J Cutan Pathol 2008; 35 Suppl 1: 83-6. ArticlePubMed
- 183. Atzmony L, Ugwu N, Zaki TD, Antaya RJ, Choate KA. Post-zygotic ACTB mutations underlie congenital smooth muscle hamartomas. J Cutan Pathol 2020; 47: 681-5. ArticlePubMedPMCPDF
- 184. Johnson MD, Jacobs AH. Congenital smooth muscle hamartoma: a report of six cases and a review of the literature. Arch Dermatol 1989; 125: 820-2. ArticlePubMed
- 185. Pitjadi TM, Chetty R, Ramdial PK, Watanabe R. EBV-associated smooth muscle tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 581-2.
- 186. Deyrup AT, Lee VK, Hill CE, et al. Epstein-Barr virus-associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: a clinicopathologic and molecular analysis of 29 tumors from 19 patients. Am J Surg Pathol 2006; 30: 75-82. ArticlePubMed
- 187. Dekate J, Chetty R. Epstein-Barr virus-associated smooth muscle tumor. Arch Pathol Lab Med 2016; 140: 718-22. ArticlePubMedPDF
- 188. Pitjadi TM, Grayson W. Epstein-Barr virus-associated smooth muscle tumour: a case series with a significant proportion of tumours showing proclivity for cutaneous soft tissues. Dermatopathology (Basel) 2019; 6: 133-46. ArticlePubMedPMCPDF
- 189. Hussein K, Rath B, Ludewig B, Kreipe H, Jonigk D. Clinico-pathological characteristics of different types of immunodeficiency-associated smooth muscle tumours. Eur J Cancer 2014; 50: 2417-24. ArticlePubMed
- 190. Issarachaikul R, Shuangshoti S, Suankratay C. Epstein-Barr virus-associated smooth muscle tumors in AIDS patients: a largest case (series). Intern Med 2014; 53: 2391-6. ArticlePubMed
- 191. Lee ES, Locker J, Nalesnik M, et al. The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation. N Engl J Med 1995; 332: 19-25. ArticlePubMed
- 192. McClain KL, Leach CT, Jenson HB, et al. Association of Epstein-Barr virus with leiomyosarcomas in young people with AIDS. N Engl J Med 1995; 332: 12-8. ArticlePubMed
- 193. Ong KW, Teo M, Lee V, et al. Expression of EBV latent antigens, mammalian target of rapamycin, and tumor suppression genes in EBV-positive smooth muscle tumors: clinical and therapeutic implications. Clin Cancer Res 2009; 15: 5350-8. ArticlePubMedPDF
- 194. Jonigk D, Laenger F, Maegel L, et al. Molecular and clinicopathological analysis of Epstein-Barr virus-associated posttransplant smooth muscle tumors. Am J Transplant 2012; 12: 1908-17. ArticlePubMed
- 195. Galeano-Piedrahita E, Rico AM, Suarez AC, Walter AL. Cutaneous smooth muscle tumors associated with Epstein-Barr virus in an adult patient with HIV. An Bras Dermatol 2021; 96: 184-7. ArticlePubMedPMC
- 196. Matin RN, Ieremia E. Cutaneous Epstein-Barr virus-associated smooth muscle tumor in immunosuppression. J Cutan Pathol 2021; 48: 325-9. ArticlePubMedPDF
- 197. Ferguson PM, Hollann TJ. Solitary circumscribed neuroma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 587-8.
- 198. Dry SM. Dermal nerve sheath myxoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 589-90.
- 199. Fetsch JF, Laskin WB, Hallman JR, Lupton GP, Miettinen M. Neurothekeoma: an analysis of 178 tumors with detailed immunohistochemical data and long-term patient follow-up information. Am J Surg Pathol 2007; 31: 1103-14. ArticlePubMed
- 200. Fetsch JF, Laskin WB, Miettinen M. Nerve sheath myxoma: a clinicopathologic and immunohistochemical analysis of 57 morphologically distinctive, S-100 protein- and GFAP-positive, myxoid peripheral nerve sheath tumors with a predilection for the extremities and a high local recurrence rate. Am J Surg Pathol 2005; 29: 1615-24. ArticlePubMed
- 201. Sheth S, Li X, Binder S, Dry SM. Differential gene expression profiles of neurothekeomas and nerve sheath myxomas by microarray analysis. Mod Pathol 2011; 24: 343-54. ArticlePubMedPDF
- 202. Zelger B, Hornick JL. Perineurioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 591-2.
- 203. Giannini C, Scheithauer BW, Jenkins RB, et al. Soft-tissue perineurioma: evidence for an abnormality of chromosome 22, criteria for diagnosis, and review of the literature. Am J Surg Pathol 1997; 21: 164-73. ArticlePubMed
- 204. Pitchford CW, Schwartz HS, Atkinson JB, Cates JM. Soft tissue perineurioma in a patient with neurofibromatosis type 2: a tumor not previously associated with the NF2 syndrome. Am J Surg Pathol 2006; 30: 1624-9. ArticlePubMed
- 205. Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol 2018; 42: 1708-14. ArticlePubMed
- 206. Brock JE, Perez-Atayde AR, Kozakewich HP, Richkind KE, Fletcher JA, Vargas SO. Cytogenetic aberrations in perineurioma: variation with subtype. Am J Surg Pathol 2005; 29: 1164-9. ArticlePubMed
- 207. Sato K, Ueda Y, Miwa S, Yokogawa A, Ozaki M, Katsuda S. Low-grade malignant soft-tissue perineurioma: interphase fluorescence in situ hybridization. Pathol Int 2008; 58: 718-22. ArticlePubMed
- 208. Shea CR, Perry A, Tetzlaff M. Neurofibroma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 593-5.
- 209. Wilson BN, John AM, Handler MZ, Schwartz RA. Neurofibromatosis type 1: new developments in genetics and treatment. J Am Acad Dermatol 2021; 84: 1667-76. ArticlePubMed
- 210. Rhodes SD, He Y, Smith A, et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum Mol Genet 2019; 28: 2752-62. ArticlePubMedPMCPDF
- 211. Yeh I, Jo VY, Perry A. Schwannoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 596-7.
- 212. Reis-Filho JS. Granular cell tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 598-600.
- 213. Pareja F, Brandes AH, Basili T, et al. Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat Commun 2018; 9: 3533.ArticlePubMedPMC
- 214. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, Kindblom LG. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol 1998; 22: 779-94. ArticlePubMed
- 215. Nasser H, Ahmed Y, Szpunar SM, Kowalski PJ. Malignant granular cell tumor: a look into the diagnostic criteria. Pathol Res Pract 2011; 207: 164-8. ArticlePubMed
- 216. Kapur P, Rakheja D, Balani JP, Roy LC, Amirkhan RH, Hoang MP. Phosphorylated histone H3, Ki-67, p21, fatty acid synthase, and cleaved caspase-3 expression in benign and atypical granular cell tumors. Arch Pathol Lab Med 2007; 131: 57-64. ArticlePubMedPDF
- 217. Nielsen GP, Chi P, Hornick JL. Malignant peripheral nerve sheath tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 603-5.
- 218. Lee W, Teckie S, Wiesner T, et al. PrC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 2014; 46: 1227-32. ArticlePubMedPMCPDF
- 219. Zhang M, Wang Y, Jones S, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 2014; 46: 1170-2. ArticlePubMedPMCPDF
- 220. Pemov A, Hansen NF, Sindiri S, et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol 2019; 21: 981-92. ArticlePubMedPMCPDF
- 221. Prieto-Granada CN, Wiesner T, Messina JL, Jungbluth AA, Chi P, Antonescu CR. Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol 2016; 40: 479-89. ArticlePubMedPMC
- 222. Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol 2016; 29: 4-13. ArticlePDF
- 223. Schaefer IM, Dong F, Garcia EP, Fletcher CD, Jo VY. Recurrent SMARCB1 inactivation in epithelioid malignant peripheral nerve sheath tumors. Am J Surg Pathol 2019; 43: 835-43. ArticlePubMedPMC
- 224. Jo VY, Fletcher CD. Smarcb1/Ini1 loss in epithelioid schwannoma: a clinicopathologic and immunohistochemical study of 65 cases. Am J Surg Pathol 2017; 41: 1013-22. ArticlePubMed
- 225. Marusic Z, Calonje JE. Dermal hyperneury/epithelial sheath neuroma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 585-6.
- 226. Ieremia E, Marusic Z, Mudaliar V, et al. Expanding the clinical spectrum of dermal hyperneury: report of nine new cases and a review of the literature. Histopathology 2019; 75: 738-45. ArticlePubMedPDF
- 227. Requena L, Grosshans E, Kutzner H, et al. Epithelial sheath neuroma: a new entity. Am J Surg Pathol 2000; 24: 190-6. ArticlePubMed
- 228. Schaffer JV, Kamino H, Witkiewicz A, McNiff JM, Orlow SJ. Mucocutaneous neuromas: an underrecognized manifestation of PTEN hamartoma-tumor syndrome. Arch Dermatol 2006; 142: 625-32. ArticlePubMed
- 229. Winkelmann RK, Carney JA. Cutaneous neuropathology in multiple endocrine neoplasia, type 2b. J Invest Dermatol 1982; 79: 307-12. ArticlePubMed
- 230. Alegria-Landa V, Jo-Velasco M, Robledo M, Requena L. Dermal hyperneury and multiple sclerotic fibromas in multiple endocrine neoplasia type 2A syndrome. JAMA Dermatol 2017; 153: 1298-301. ArticlePubMed
- 231. Rhee DY, Chung WK, Chang SE, Lee MW, Choi JH, Moon KC. A case of cutaneous nerve hypertrophy: association with multiple endocrine neoplasia type 2 or neurofibromatosis type 2? J Cutan Pathol 2008; 35: 1156-8. ArticlePubMed
- 232. Starink TM, van der Veen JP, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet 1986; 29: 222-33. ArticlePubMed
- 233. Ohtake Y, Hayat U, Li S. Pten inhibition and axon regeneration and neural repair. Neural Regen Res 2015; 10: 1363-8. ArticlePubMedPMC
- 234. Noparstak M, Zaycosky M, Saeed S. Epithelial sheath neuroma with extension to the subcutis. J Cutan Pathol 2018; 45: 705-7. ArticlePDF
- 235. Stemmer-Rachamimov AO, Hornick JL. Hybrid nerve sheath tumours. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 601-2.
- 236. Hornick JL, Bundock EA, Fletcher CD. Hybrid schwannoma/perineurioma: clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am J Surg Pathol 2009; 33: 1554-61. ArticlePubMed
- 237. Feany MB, Anthony DC, Fletcher CD. Nerve sheath tumours with hybrid features of neurofibroma and schwannoma: a conceptual challenge. Histopathology 1998; 32: 405-10. ArticlePubMedPDF
- 238. Michal M, Kazakov DV, Belousova I, Bisceglia M, Zamecnik M, Mukensnabl P. A benign neoplasm with histopathological features of both schwannoma and retiform perineurioma (benign schwannoma-perineurioma): a report of six cases of a distinctive soft tissue tumor with a predilection for the fingers. Virchows Arch 2004; 445: 347-53. ArticlePubMedPDF
- 239. Harder A, Wesemann M, Hagel C, et al. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am J Surg Pathol 2012; 36: 702-9. ArticlePubMed
- 240. MacCollin M, Chiocca EA, Evans DG, et al. Diagnostic criteria for schwannomatosis. Neurology 2005; 64: 1838-45. ArticlePubMed
- 241. Inatomi Y, Ito T, Nagae K, et al. Hybrid perineurioma-neurofibroma in a patient with neurofibromatosis type 1, clinically mimicking malignant peripheral nerve sheath tumor. Eur J Dermatol 2014; 24: 412-3. ArticlePubMed
- 242. Kacerovska D, Michal M, Kuroda N, et al. Hybrid peripheral nerve sheath tumors, including a malignant variant in type 1 neurofibromatosis. Am J Dermatopathol 2013; 35: 641-9. ArticlePubMed
- 243. Dickson BC, Antonescu CR, Demicco EG, et al. Hybrid schwannoma-perineurioma frequently harbors VGLL3 rearrangement. Mod Pathol 2021; 34: 1116-24. ArticlePubMedPMCPDF
- 244. Agaimy A. Microscopic intraneural perineurial cell proliferations in patients with neurofibromatosis type 1. Ann Diagn Pathol 2014; 18: 95-8. ArticlePubMed
- 245. Requena L, Hornick JL. Cellular neurothekeoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 606-7.
- 246. Barnhill RL. Nerve sheath myxoma (neurothekeoma). J Cutan Pathol 1994; 21: 91-3. ArticlePubMed
- 247. Abdaljaleel M, North JP. Positive MITF and NKI/C3 expression in cellular neurothekeoma and dermatofibroma. Appl Immunohistochem Mol Morphol 2021; 29: 440-5. ArticlePubMed
- 248. Argenyi ZB, LeBoit PE, Santa Cruz D, Swanson PE, Kutzner H. Nerve sheath myxoma (neurothekeoma) of the skin: light microscopic and immunohistochemical reappraisal of the cellular variant. J Cutan Pathol 1993; 20: 294-303. ArticlePubMed
- 249. Wang AR, May D, Bourne P, Scott G. PGP9.5: a marker for cellular neurothekeoma. Am J Surg Pathol 1999; 23: 1401-7. ArticlePubMed
- 250. Fullen DR, Lowe L, Su LD. Antibody to S100A6 protein is a sensitive immunohistochemical marker for neurothekeoma. J Cutan Pathol 2003; 30: 118-22. ArticlePubMedPDF
- 251. Suarez A, High WA. Immunohistochemical analysis of KBA.62 in 18 neurothekeomas: a potential marker for differentiating neurothekeoma, but a marker that may lead to confusion with melanocytic tumors. J Cutan Pathol 2014; 41: 36-41. ArticlePubMed
- 252. Cesinaro AM, Piana S, Paganelli A, Pedroni G, Santandrea G, Maiorana A. Prame expression in cellular neurothekeoma: a study of 11 cases. J Cutan Pathol 2022; 49: 338-42. ArticlePubMedPDF
- 253. Lazar AJ, Yeh I. Non-neural granular cell tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 610-1.
- 254. Cohen JN, Yeh I, Jordan RC, et al. Cutaneous non-neural granular cell tumors harbor recurrent ALK gene fusions. Am J Surg Pathol 2018; 42: 1133-42. ArticlePubMed
- 255. Beer TW, Calonje JE. Atypical fibroxanthoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 618-20.
- 256. Griewank KG, Wiesner T, Murali R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma harbor frequent NOTCH1/2 and FAT1 mutations and similar DNA copy number alteration profiles. Mod Pathol 2018; 31: 418-28. ArticlePubMedPDF
- 257. Koelsche C, Stichel D, Griewank KG, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res 2019; 9: 2.ArticlePubMedPMCPDF
- 258. Ak M, Kahraman A, Arnold FM, et al. Clinicopathological and genomic profiles of atypical fibroxanthoma and pleomorphic dermal sarcoma identify overlapping signatures with a high mutational burden. Genes (Basel) 2021; 12: 974.ArticlePubMedPMC
- 259. Harding-Jackson N, Sangueza M, Mackinnon A, Suster S, Plaza JA. Spindle cell atypical fibroxanthoma: myofibroblastic differentiation represents a diagnostic pitfall in this variant of AFX. Am J Dermatopathol 2015; 37: 509-14. ArticlePubMed
- 260. Tardio JC, Pinedo F, Aramburu JA, et al. Clear cell atypical fibroxanthoma: clinicopathological study of 6 cases and review of the literature with special emphasis on the differential diagnosis. Am J Dermatopathol 2016; 38: 586-92. ArticlePubMed
- 261. Diaz-Cascajo C, Borghi S, Bonczkowitz M. Pigmented atypical fibroxanthoma. Histopathology 1998; 33: 537-41. ArticlePubMedPDF
- 262. Dummer R, Luzar B, Miller K. Pleomorphic dermal sarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 623-4.
- 263. Patel RM, Le Loarer F, Nielsen TO, Oda Y. Epithelioid sarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 625-6.
- 264. Kohashi K, Yamamoto H, Yamada Y, et al. Swi/Snf chromatin-remodeling complex status in SMARCB1/INI1-preserved epithelioid sarcoma. Am J Surg Pathol 2018; 42: 312-8. ArticlePubMed
- 265. Jamshidi F, Bashashati A, Shumansky K, et al. The genomic landscape of epithelioid sarcoma cell lines and tumours. J Pathol 2016; 238: 63-73. ArticlePubMed
- 266. Busam KJ, Fritchie KJ, Luzar B. Dermal clear cells sarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 629-30.
- 267. Raghavan SS, Wang JY, Toland A, et al. Diffuse PRAME expression is highly specific for malignant melanoma in the distinction from clear cell sarcoma. J Cutan Pathol 2020; 47: 1226-8. ArticlePubMedPDF
- 268. Kline N, Menge TD, Hrycaj SM, et al. Prame expression in challenging dermal melanocytic neoplasms and soft tissue tumors with melanocytic differentiation. Am J Dermatopathol 2022; 44: 404-10. ArticlePubMed
- 269. Patel RM, de Alava E, Scolyer RA. Ewing sarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 631-2.
- 270. Choi EY, Gardner JM, Lucas DR, McHugh JB, Patel RM. Ewing sarcoma. Semin Diagn Pathol 2014; 31: 39-47. ArticlePubMed
- 271. Hasegawa SL, Davison JM, Rutten A, Fletcher JA, Fletcher CD. Primary cutaneous Ewing's sarcoma: immunophenotypic and molecular cytogenetic evaluation of five cases. Am J Surg Pathol 1998; 22: 310-8. ArticlePubMed
- 272. Machado I, Llombart B, Calabuig-Farinas S, Llombart-Bosch A. Superficial Ewing's sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol 2011; 38: 636-43. ArticlePubMed
- 273. Requena L, Hornick JL. Epithelioid fibrous histiocytoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 608-9.
- 274. Glusac EJ, Barr RJ, Everett MA, Pitha J, Santa Cruz DJ. Epithelioid cell histiocytoma: a report of 10 cases including a new cellular variant. Am J Surg Pathol 1994; 18: 583-90. PubMed
- 275. Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology 1994; 24: 123-9. ArticlePubMed
- 276. Doyle LA, Marino-Enriquez A, Fletcher CD, Hornick JL. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol 2015; 28: 904-12. ArticlePubMedPDF
- 277. Walther C, Hofvander J, Nilsson J, et al. Gene fusion detection in formalin-fixed paraffin-embedded benign fibrous histiocytomas using fluorescence in situ hybridization and RNA sequencing. Lab Invest 2015; 95: 1071-6. ArticlePubMedPDF
- 278. Jedrych J, Nikiforova M, Kennedy TF, Ho J. Epithelioid cell histiocytoma of the skin with clonal ALK gene rearrangement resulting in VCL-ALK and SQSTM1-ALK gene fusions. Br J Dermatol 2015; 172: 1427-9. ArticlePubMed
- 279. Dickson BC, Swanson D, Charames GS, Fletcher CD, Hornick JL. Epithelioid fibrous histiocytoma: molecular characterization of ALK fusion partners in 23 cases. Mod Pathol 2018; 31: 753-62. ArticlePubMedPDF
- 280. Kazakov DV, Kyrpychova L, Martinek P, et al. ALK gene fusions in epithelioid fibrous histiocytoma: a study of 14 cases, with new histopathological findings. Am J Dermatopathol 2018; 40: 805-14. ArticlePubMed
- 281. Georgantzoglou N, Green D, Winnick KN, et al. Molecular investigation of ALK-rearranged epithelioid fibrous histiocytomas identifies CLTC as a novel fusion partner and evidence of fusion-independent transcription activation. Genes Chromosomes Cancer 2022; 61: 471-80. ArticlePubMed
- 282. Wright GR, Archibald CW, Fontaine D, Dakin-Hache K, Walsh NM. Epithelioid fibrous histiocytoma with chondroblastoma-like features: a report of a rare entity and discussion of related diagnostic challenges. Am J Dermatopathol 2022; 44: e11-5. ArticlePubMed
- 283. Murigu T, Bhatt N, Miller K, Palmer A, Melegh Z. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology 2018; 72: 1233-6. ArticlePDF
- 284. Kazlouskaya V, Ho J, Jedrych J, Karunamurthy A. Spindle cell variant of epithelioid cell histiocytoma (spindle cell histiocytoma) with ALK gene fusions: cases series and review of the literature. J Cutan Pathol 2021; 48: 837-41. ArticlePubMedPDF
- 285. Moayed-Alaei L, Vargas AC, Adybeik D, Maclean F, Moir D. Analyzing the morphological spectrum of epithelioid fibrous histiocytoma and the immunohistochemical performance of the ALK D5F3 and ALK1 clones. Hum Pathol 2022; 120: 46-56. ArticlePubMed
- 286. Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol 2011; 38: 697-703. ArticlePubMed
- 287. Doyle LA, Argani P, Hornick JL. PEComa. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 612-3.
- 288. Liegl B, Hornick JL, Fletcher CD. Primary cutaneous PEComa: distinctive clear cell lesions of skin. Am J Surg Pathol 2008; 32: 608-14. ArticlePubMed
- 289. Stuart LN, Tipton RG, DeWall MR, et al. Primary cutaneous perivascular epithelioid cell tumor (PEComa): five new cases and review of the literature. J Cutan Pathol 2017; 44: 713-21. ArticlePubMedPDF
- 290. Greveling K, Winnepenninckx VJL, Nagtzaam IF, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol 2013; 69: e262-4. ArticlePubMed
- 291. Odono EI, Tan KB, Tay SY, Lee VK. Cutaneous "fibroma-like" perivascular epithelioid cell tumor: a case report and review of literature. J Cutan Pathol 2020; 47: 548-53. ArticlePubMedPDF
- 292. Pan CC, Chung MY, Ng KF, et al. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol 2008; 214: 387-93. ArticlePubMedPDF
- 293. Agaram NP, Sung YS, Zhang L, et al. Dichotomy of genetic abnormalities in PEComas with therapeutic implications. Am J Surg Pathol 2015; 39: 813-25. ArticlePubMedPMC
- 294. Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 2005; 29: 1558-75. ArticlePubMed
- 295. Doyle LA, Hornick JL, Fletcher CD. Pecoma of the gastrointestinal tract: clinicopathologic study of 35 cases with evaluation of prognostic parameters. Am J Surg Pathol 2013; 37: 1769-82. ArticlePubMed
- 296. Bennett JA, Braga AC, Pinto A, et al. Uterine PEComas: a morphologic, immunohistochemical, and molecular analysis of 32 tumors. Am J Surg Pathol 2018; 42: 1370-83. ArticlePubMedPMC
- 297. Cole DW, Menge TD, Renati S, et al. Primary cutaneous malignant perivascular epithelioid cell tumor: case of a rare tumor with review of the literature. J Cutan Pathol 2021; 48: 1088-93. ArticlePubMedPDF
- 298. Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol 2010; 41: 1-15. ArticlePubMed
- 299. Hornick JL, Fletcher CD. Sclerosing PEComa: clinicopathologic analysis of a distinctive variant with a predilection for the retroperitoneum. Am J Surg Pathol 2008; 32: 493-501. ArticlePubMed
- 300. Rekhi B, Chen G. Angiomatoid fibrous histiocytoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 614-5.
- 301. Fanburg-Smith JC, Miettinen M. Angiomatoid "malignant" fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol 1999; 30: 1336-43. ArticlePubMed
- 302. Costa MJ, Weiss SW. Angiomatoid malignant fibrous histiocytoma: a follow-up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol 1990; 14: 1126-32. PubMed
- 303. Antonescu CR, Dal Cin P, Nafa K, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2007; 46: 1051-60. ArticlePubMed
- 304. Rekhi B, Adamane S, Ghodke K, Desai S, Jambhekar NA. Angiomatoid fibrous histiocytoma: clinicopathological spectrum of five cases, including EWSR1-CREB1 positive result in a single case. Indian J Pathol Microbiol 2016; 59: 148-52. ArticlePubMed
- 305. Rossi S, Szuhai K, Ijszenga M, et al. EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res 2007; 13: 7322-8. ArticlePubMedPDF
- 306. Hallor KH, Mertens F, Jin Y, et al. Fusion of the EWSR1 and ATF1 genes without expression of the MITF-M transcript in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2005; 44: 97-102. ArticlePubMed
- 307. Raddaoui E, Donner LR, Panagopoulos I. Fusion of the FUS and ATF1 genes in a large, deep-seated angiomatoid fibrous histiocytoma. Diagn Mol Pathol 2002; 11: 157-62. ArticlePubMed
- 308. Waters BL, Panagopoulos I, Allen EF. Genetic characterization of angiomatoid fibrous histiocytoma identifies fusion of the FUS and ATF-1 genes induced by a chromosomal translocation involving bands 12q13 and 16p11. Cancer Genet Cytogenet 2000; 121: 109-16. ArticlePubMed
- 309. Thway K, Fisher C. Angiomatoid fibrous histiocytoma: the current status of pathology and genetics. Arch Pathol Lab Med 2015; 139: 674-82. ArticlePubMedPDF
- 310. Kao YC, Lan J, Tai HC, et al. Angiomatoid fibrous histiocytoma: clinicopathological and molecular characterisation with emphasis on variant histomorphology. J Clin Pathol 2014; 67: 210-5. ArticlePubMed
- 311. Hasegawa T, Seki K, Ono K, Hirohashi S. Angiomatoid (malignant) fibrous histiocytoma: a peculiar low-grade tumor showing immunophenotypic heterogeneity and ultrastructural variations. Pathol Int 2000; 50: 731-8. ArticlePubMedPDF
- 312. Schaefer IM, Fletcher CD. Myxoid variant of so-called angiomatoid "malignant fibrous histiocytoma": clinicopathologic characterization in a series of 21 cases. Am J Surg Pathol 2014; 38: 816-23. ArticlePubMed
- 313. Chen G, Folpe AL, Colby TV, et al. Angiomatoid fibrous histiocytoma: unusual sites and unusual morphology. Mod Pathol 2011; 24: 1560-70. ArticlePubMedPDF
- 314. Berklite L, John I, Ranganathan S, Parafioriti A, Alaggio R. Sox9 immunohistochemistry in the distinction of angiomatoid fibrous histiocytoma from histologic mimics: diagnostic utility and pitfalls. Appl Immunohistochem Mol Morphol 2020; 28: 635-40. ArticlePubMed
- 315. Suurmeijer AJ, Davis JL. NTRK-rearranged spindle cell neoplasm. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 616-7.
- 316. Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol 2016; 40: 1407-16. ArticlePubMedPMC
- 317. Antonescu CR, Dickson BC, Swanson D, et al. Spindle cell tumors with RET gene fusions exhibit a morphologic spectrum akin to tumors with NTRK gene fusions. Am J Surg Pathol 2019; 43: 1384-91. ArticlePubMedPMC
- 318. Kao YC, Suurmeijer AJ, Argani P, et al. Soft tissue tumors characterized by a wide spectrum of kinase fusions share a lipofibromatosis-like neural tumor pattern. Genes Chromosomes Cancer 2020; 59: 575-83. ArticlePubMedPMCPDF
- 319. Davis JL, Lockwood CM, Stohr B, et al. Expanding the spectrum of pediatric NTRK-rearranged mesenchymal tumors. Am J Surg Pathol 2019; 43: 435-45. ArticlePubMed
- 320. Hung YP, Fletcher CD, Hornick JL. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology 2018; 73: 634-44. ArticlePubMedPDF
- 321. Rekhi B, Folpe AL, Yu L. Superficial CD34-positive fibroblastic tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 621-2.
- 322. Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol 2014; 27: 294-302. ArticlePubMedPDF
- 323. Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology 2017; 70: 394-401. ArticlePubMedPDF
- 324. Perret R, Michal M, Carr RA, et al. Superficial CD34-positive fibroblastic tumor and PRDM10-rearranged soft tissue tumor are overlapping entities: a comprehensive study of 20 cases. Histopathology 2021; 79: 810-25. ArticlePubMedPDF
- 325. Puls F, Carter JM, Pillay N, et al. Overlapping morphological, immunohistochemical and genetic features of superficial CD34-positive fibroblastic tumor and PRDM10-rearranged soft tissue tumor. Mod Pathol 2022; 35: 767-76. ArticlePubMedPDF
- 326. Ding L, Xu WJ, Tao XY, Zhang L, Cai ZG. Clinicopathological features of superficial CD34-positive fibroblastic tumor. World J Clin Cases 2021; 9: 2739-50. ArticlePubMedPMC
- 327. Mao X, Sun YY, Deng ML, Ma T, Yu L. Superficial CD34-positive fibroblastic tumor: report of two cases and review of literature. Int J Clin Exp Pathol 2020; 13: 38-43. PubMedPMC
- 328. Puls F, Pillay N, Fagman H, et al. PRDM10-rearranged soft tissue tumor: a clinicopathologic study of 9 cases. Am J Surg Pathol 2019; 43: 504-13. ArticlePubMed
- 329. de la Fouchardiere A, Ko JS. CRTC1::TRIM11 cutaneous tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 627-8.
- 330. de la Fouchardiere A, Hanna J. MITF pathway–activated melanocytic tumours. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. Lyon: IARC Press, 2025; 136-7.
- 331. Hanna J, Ko JS, Billings SD, et al. Cutaneous melanocytic tumor with CRTC1::TRIM11 translocation: an emerging entity analyzed in a series of 41 cases. Am J Surg Pathol 2022; 46: 1457-66. ArticlePubMed
- 332. Cellier L, Perron E, Pissaloux D, et al. Cutaneous melanocytoma with CRTC1-TRIM11 fusion: report of 5 cases resembling clear cell sarcoma. Am J Surg Pathol 2018; 42: 382-91. ArticlePubMed
- 333. Ko JS, Wang L, Billings SD, et al. CRTC1-TRIM11 fusion defined melanocytic tumors: a series of four cases. J Cutan Pathol 2019; 46: 810-8. ArticlePubMedPDF
- 334. Bontoux C, Baroudjian B, Le Maignan C, et al. CRTC1-TRIM11 fusion in a case of metastatic clear cell sarcoma: are crtc1-trim11 fusion-bearing tumors melanocytomas or clear cell sarcomas? Am J Surg Pathol 2019; 43: 861-3. ArticlePubMed
- 335. Kashima J, Motoi T, Nishimaki M, et al. A case report of cutaneous melanocytoma with CRTC1-TRIM11 fusion: is CMCT distinct from clear cell sarcoma of soft tissue? Pathol Int 2019; 69: 496-501. ArticlePubMedPDF
- 336. Parra O, Bridge JA, Busam KJ, Shalin SC, Linos K. Dermal melanocytic tumor with CRTC1-TRIM11 fusion: report of two additional cases with review of the literature of an emerging entity. J Cutan Pathol 2021; 48: 915-24. ArticlePubMedPDF
- 337. Vest BE, Harview CL, Liu V, et al. Cutaneous melanocytic tumor with CRTC1::TRIM11 fusion and prominent epidermal involvement: a case report. J Cutan Pathol 2022; 49: 1025-30. ArticlePubMedPMCPDF
- 338. Yang L, Yin Z, Wei J, et al. Cutaneous melanocytic tumour with CRTC1::TRIM11 fusion in a case with recurrent local lymph node and distant pulmonary metastases at early stage: aggressive rather than indolent? Histopathology 2023; 82: 368-71. ArticlePubMedPDF
- 339. Iourgenko V, Zhang W, Mickanin C, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci U S A 2003; 100: 12147-52. ArticlePubMedPMC
- 340. Conkright MD, Canettieri G, Screaton R, et al. Torcs: transducers of regulated CREB activity. Mol Cell 2003; 12: 413-23. ArticlePubMed
- 341. Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest 2017; 97: 649-56. ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
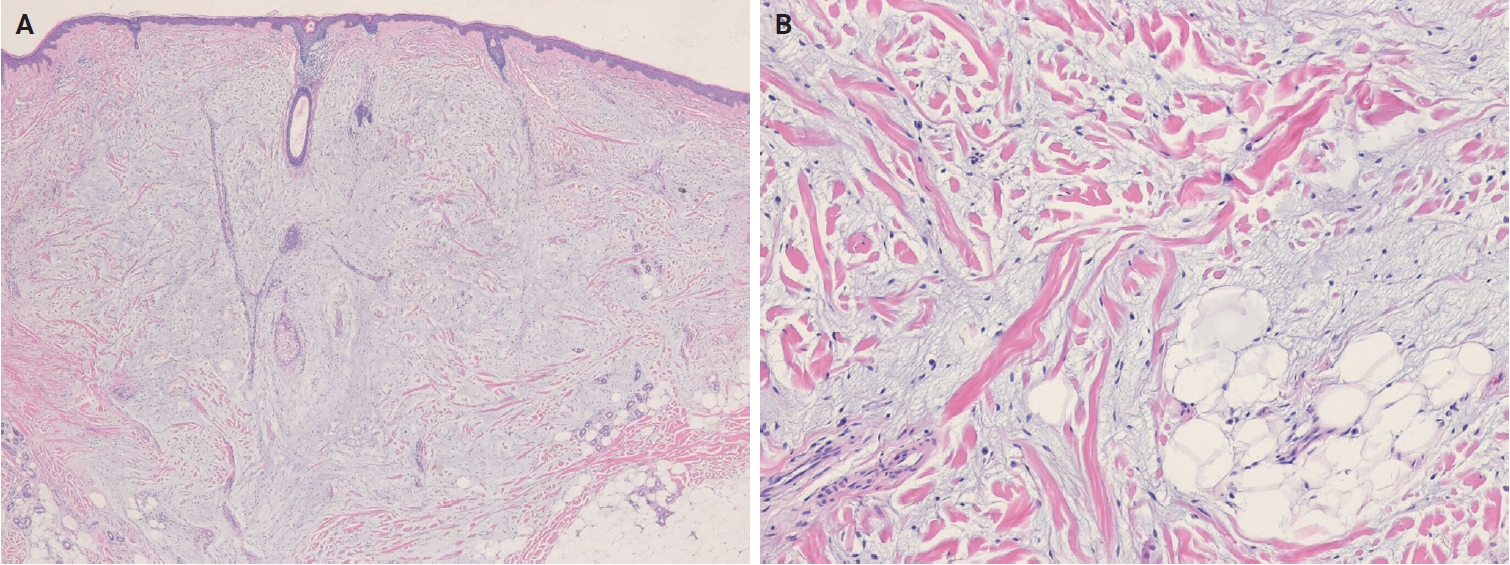
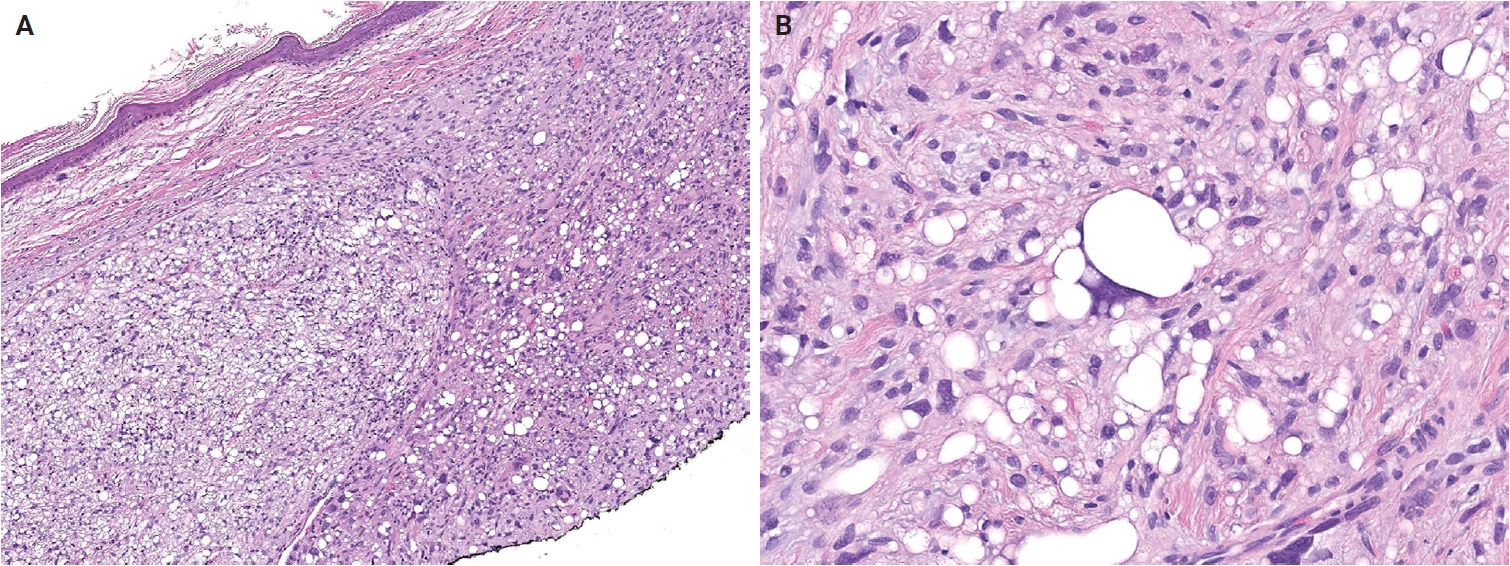
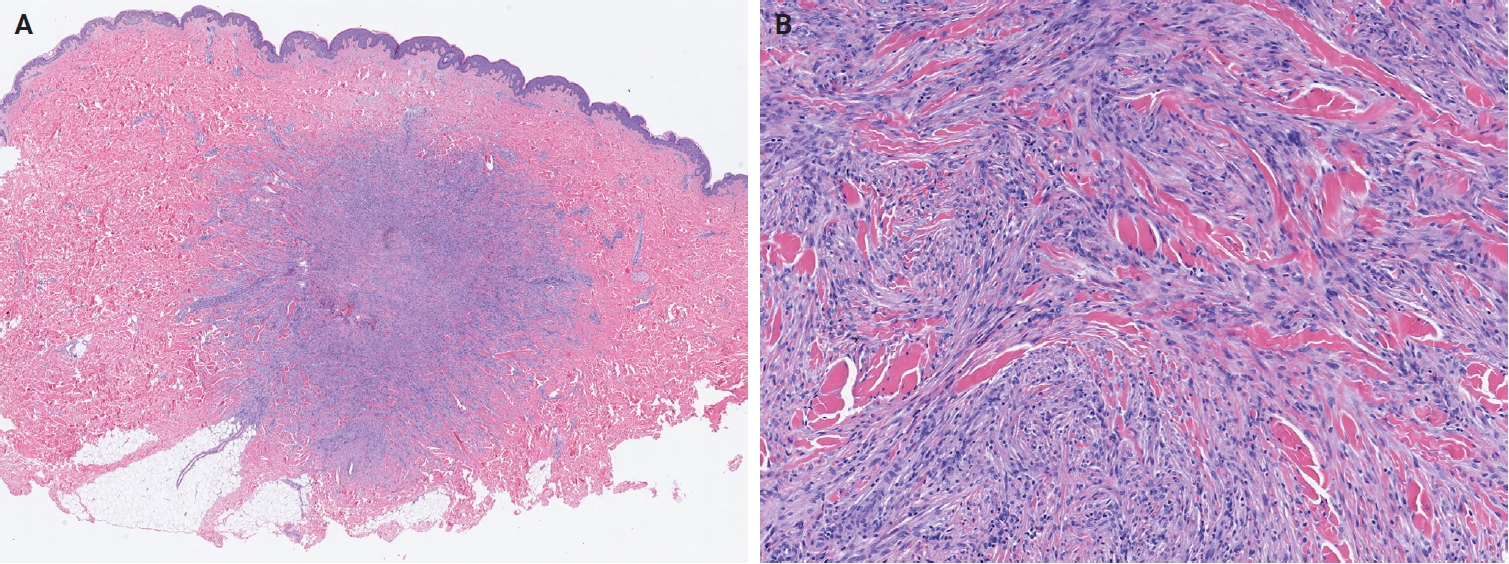
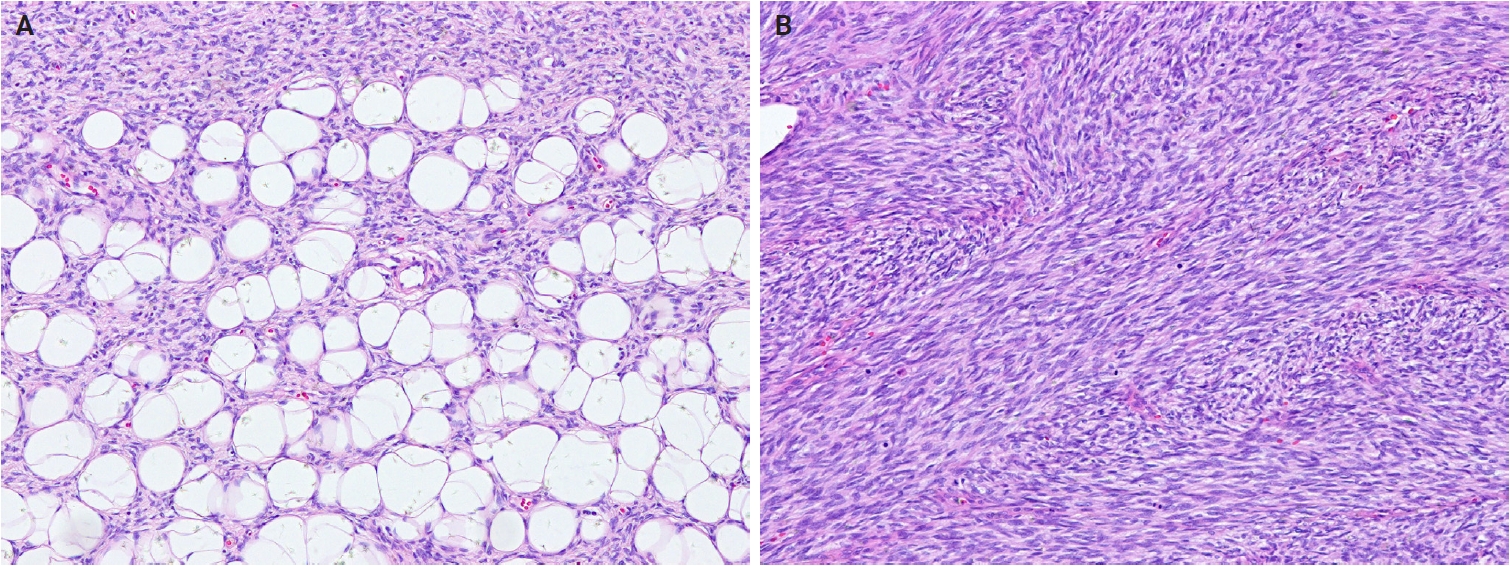
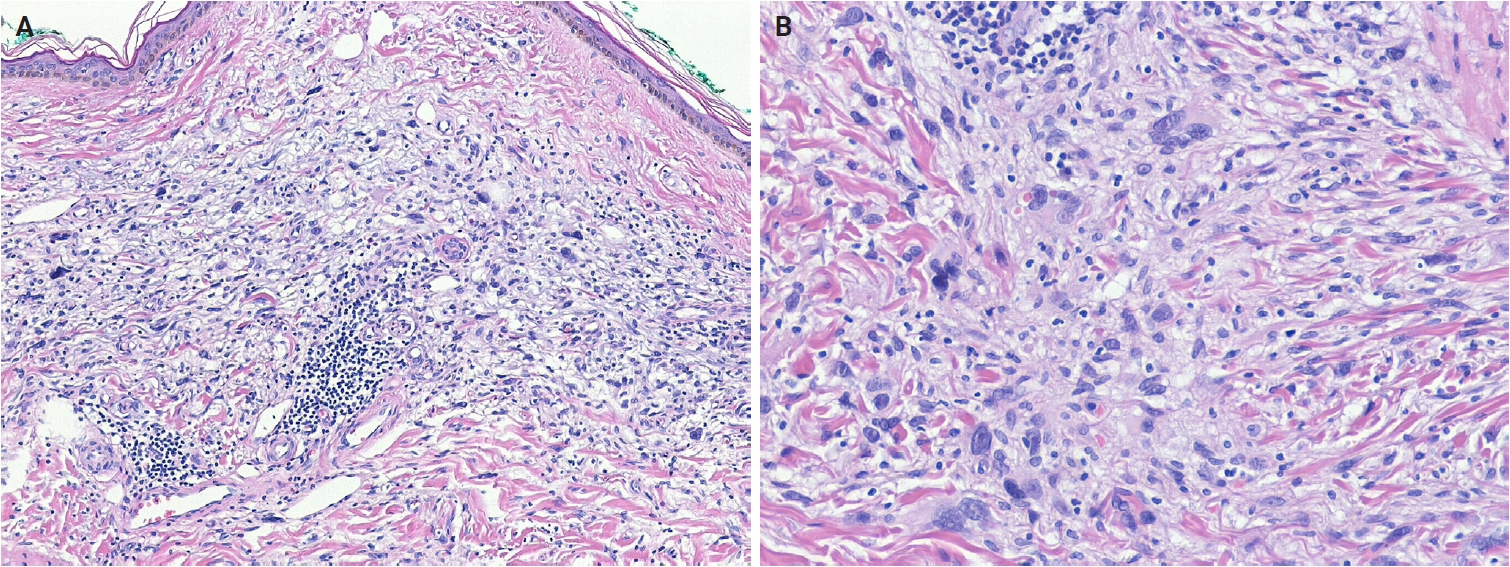
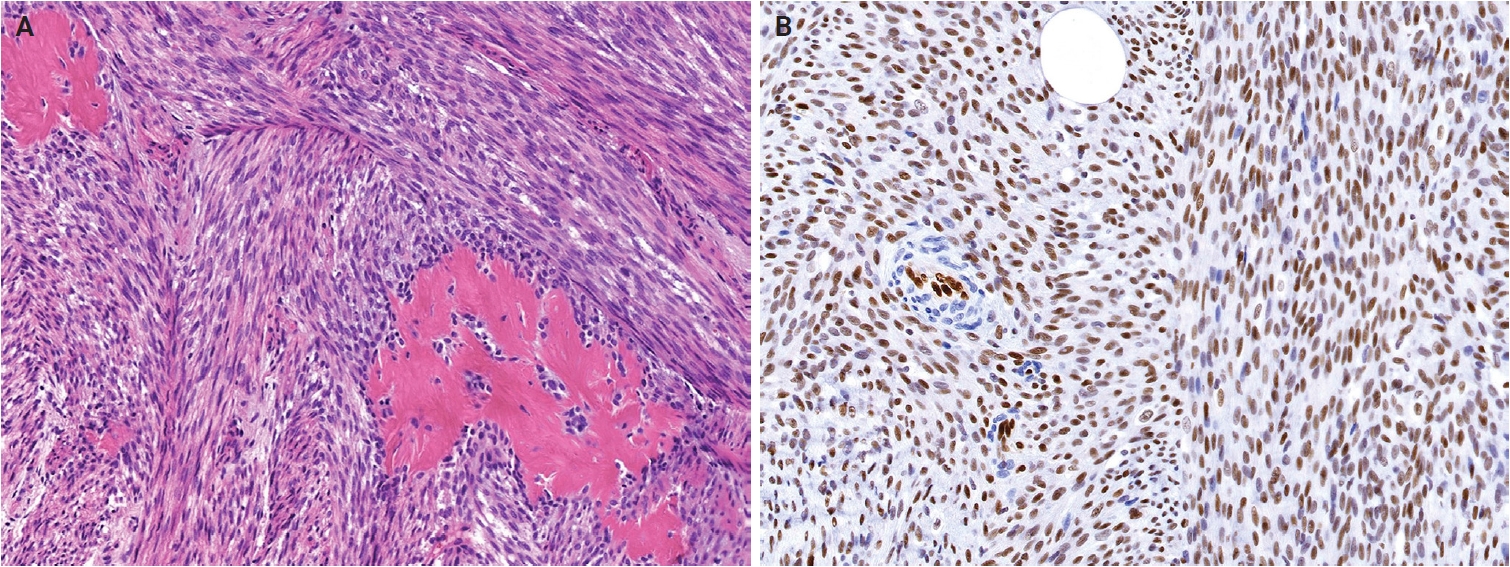
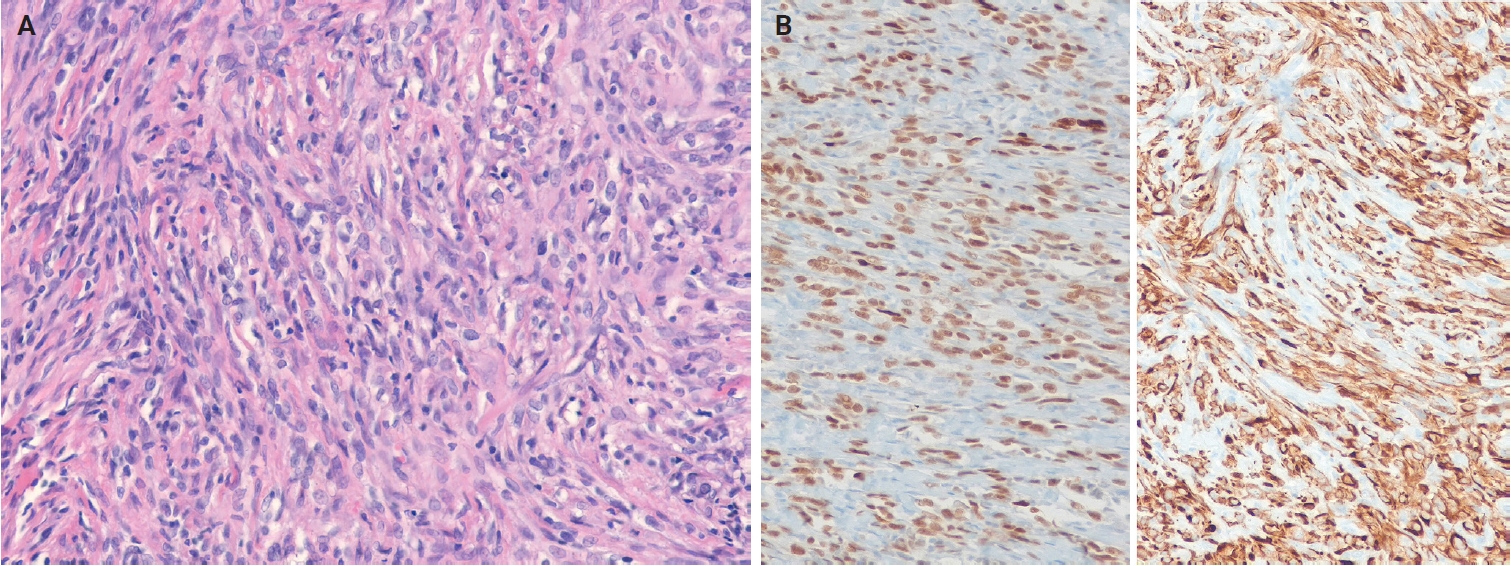
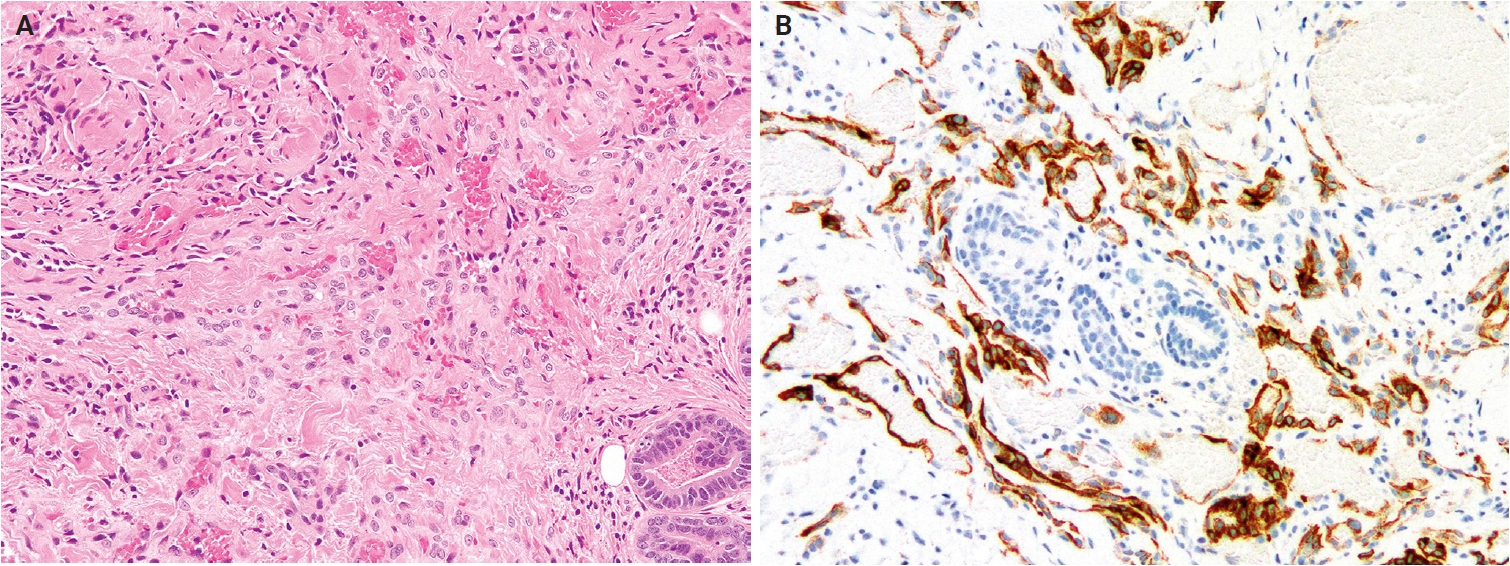
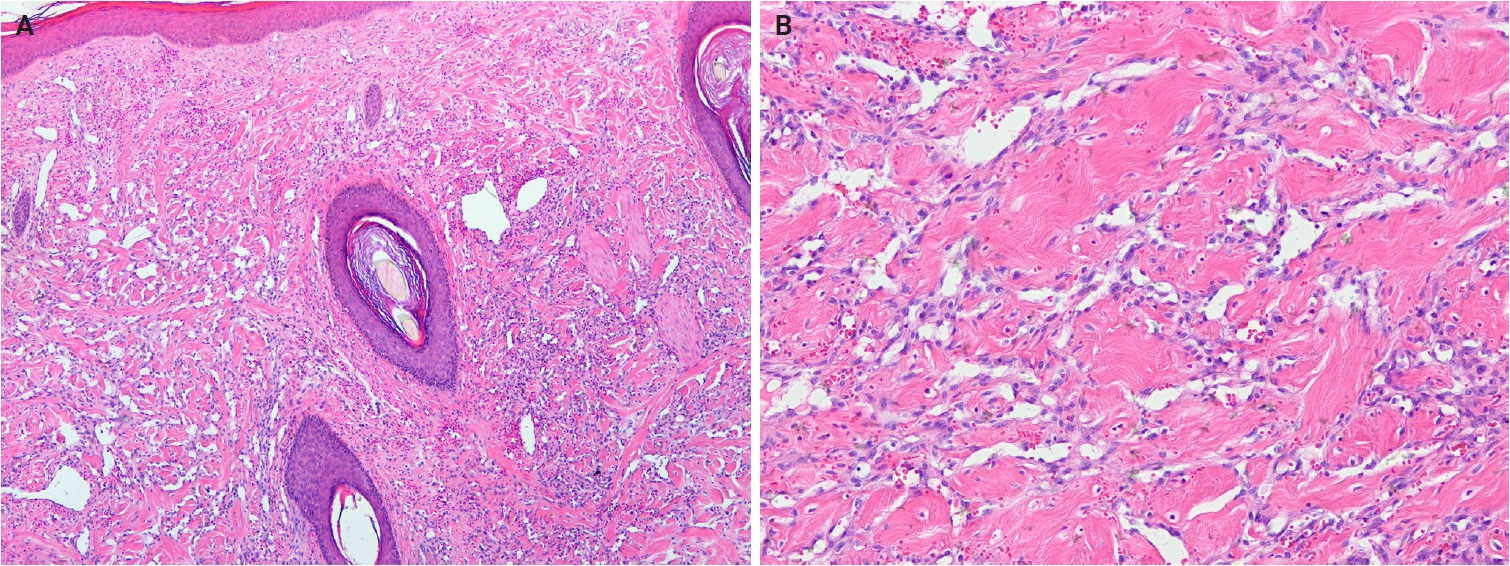
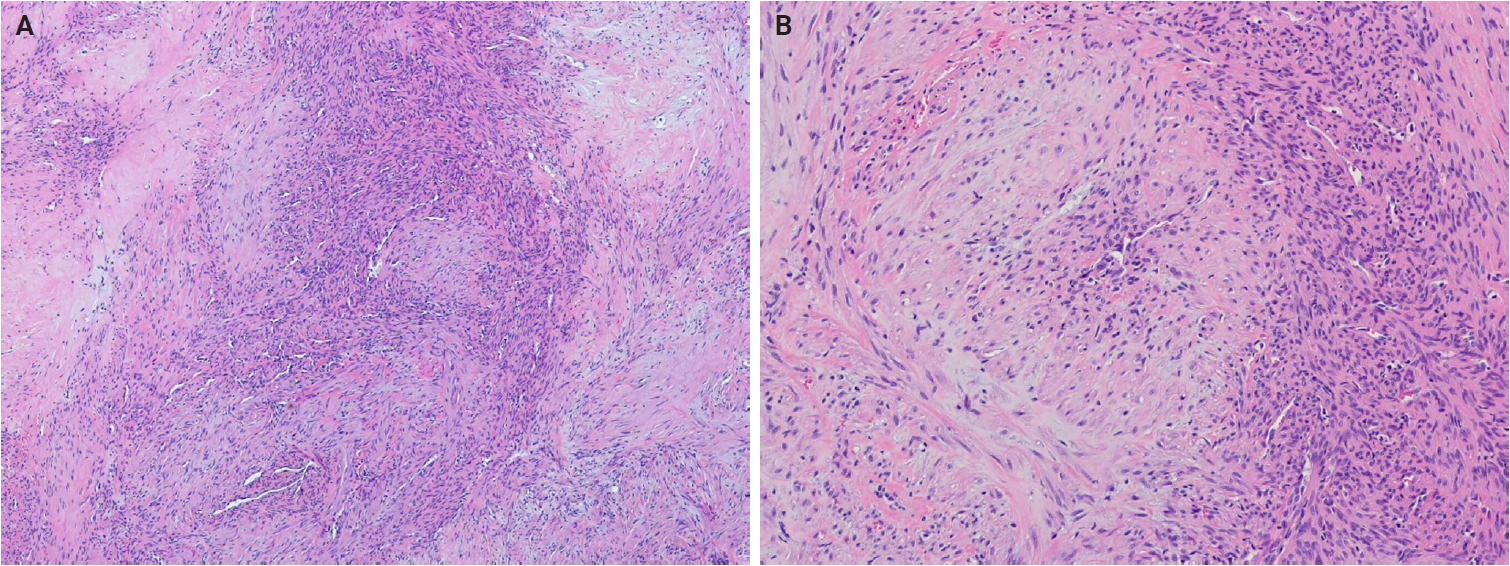
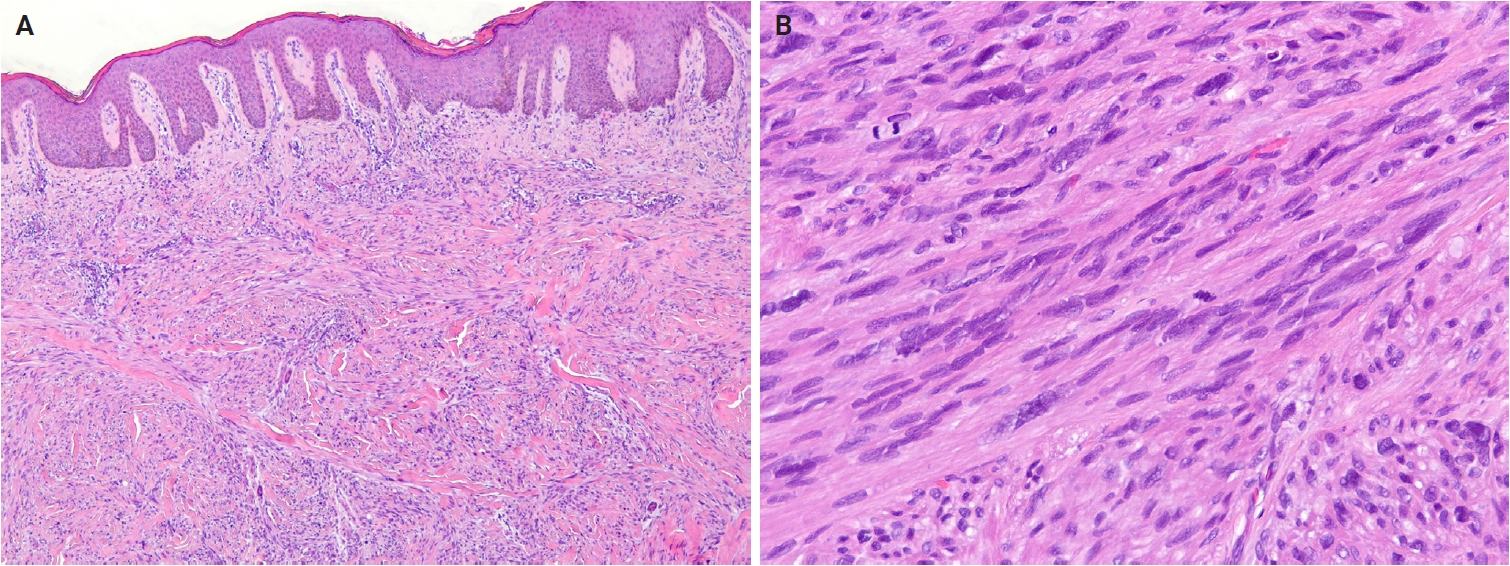
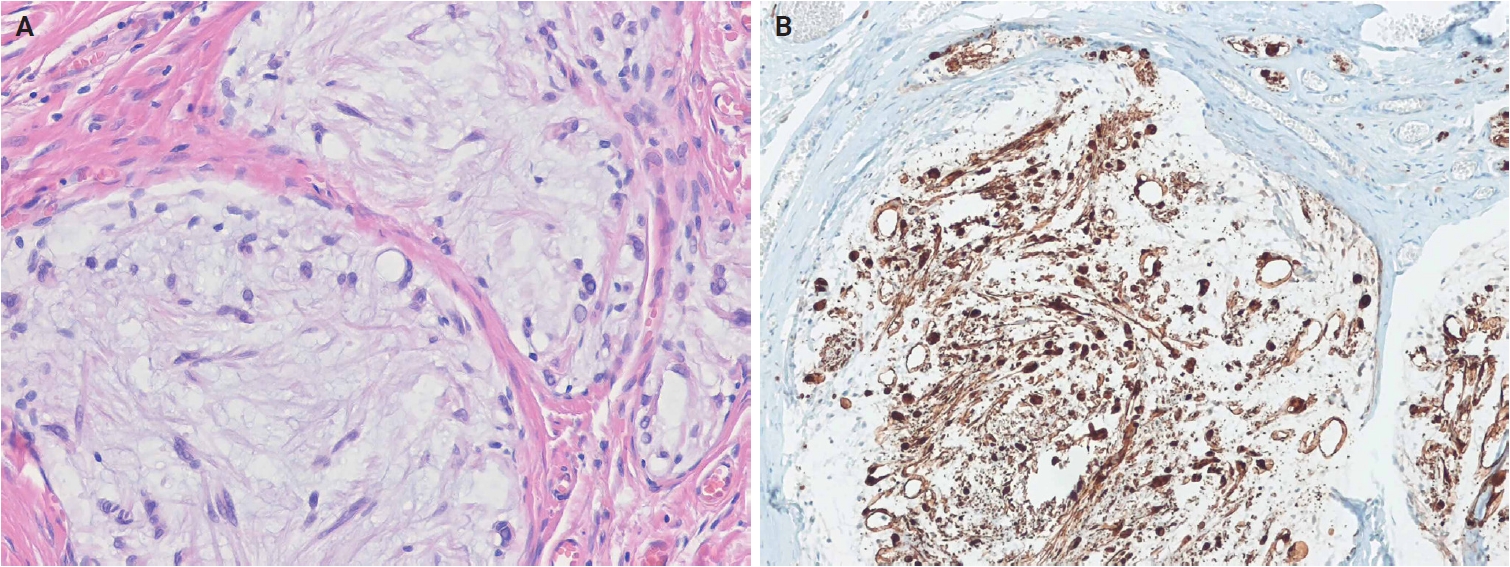
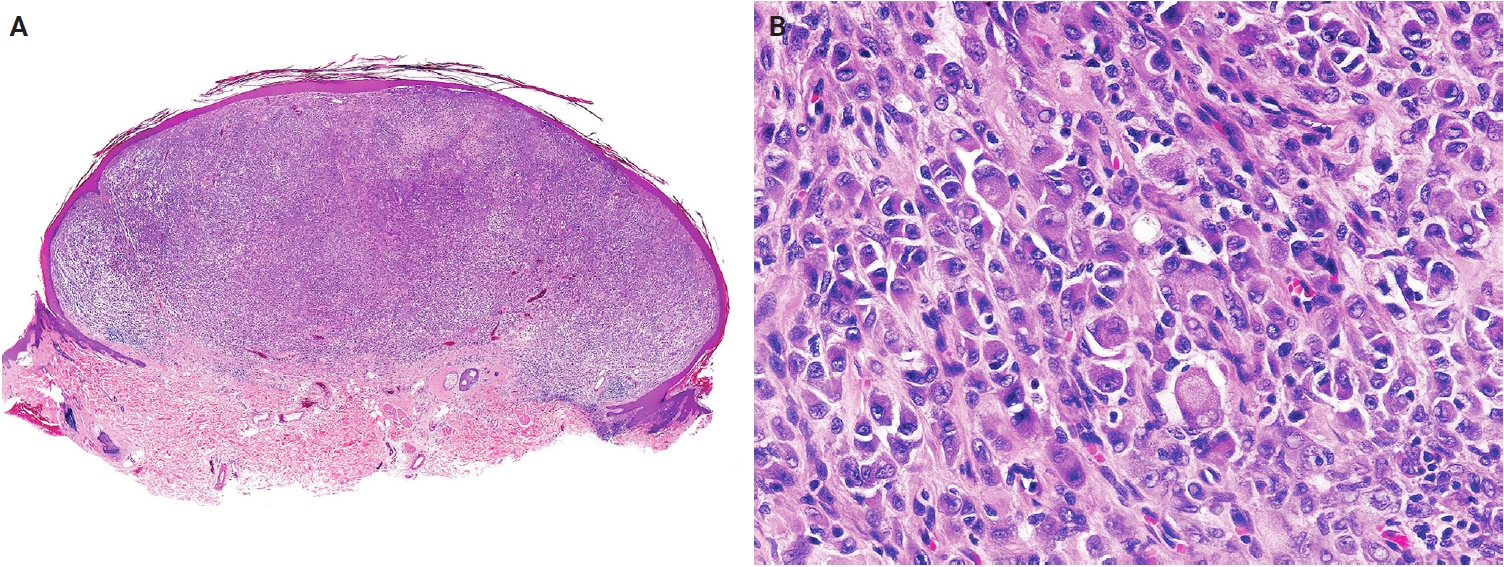
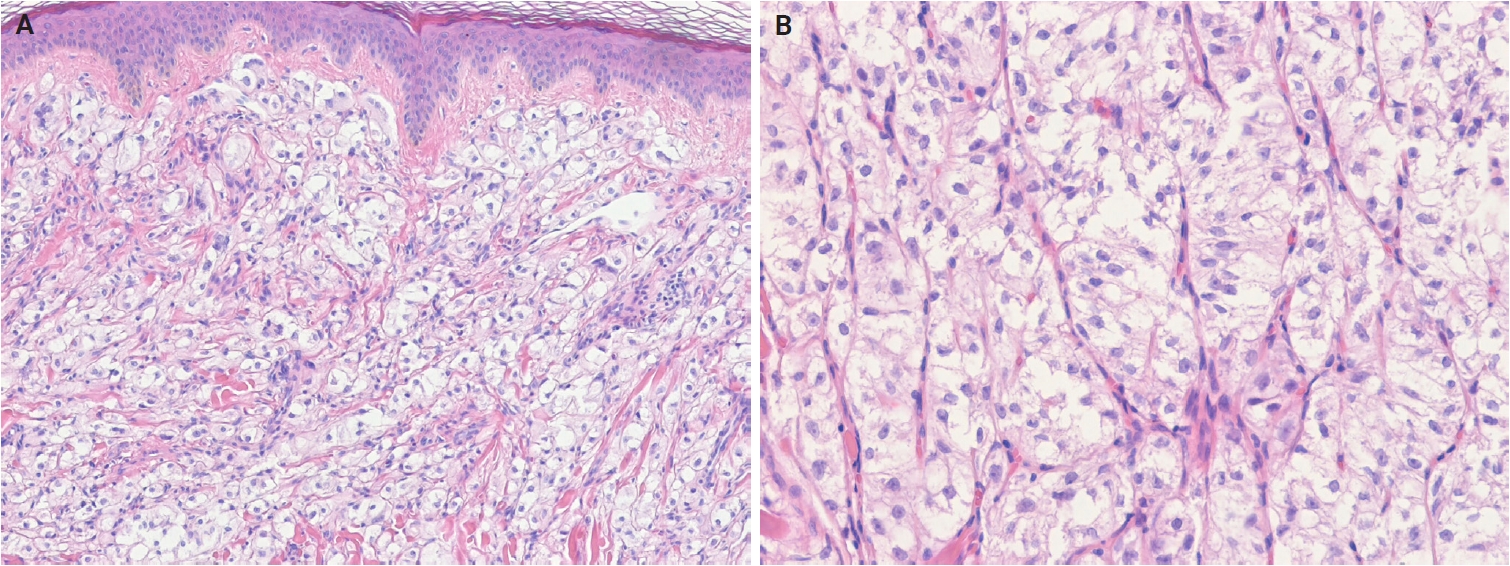
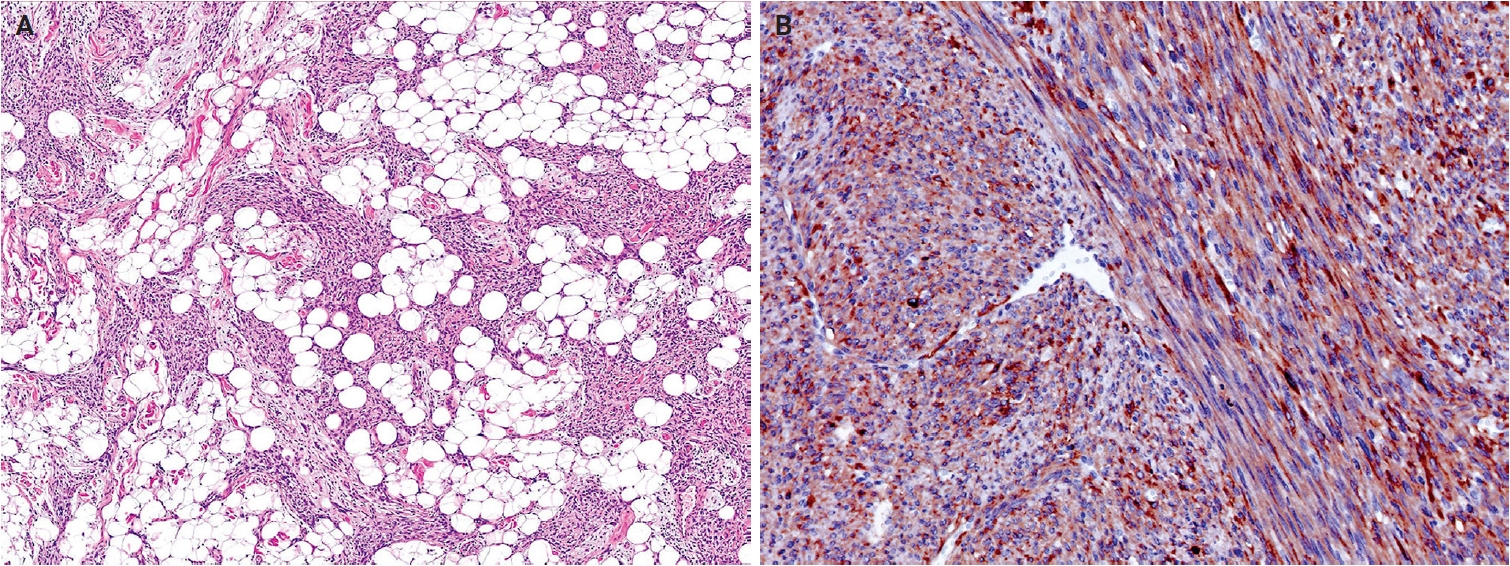
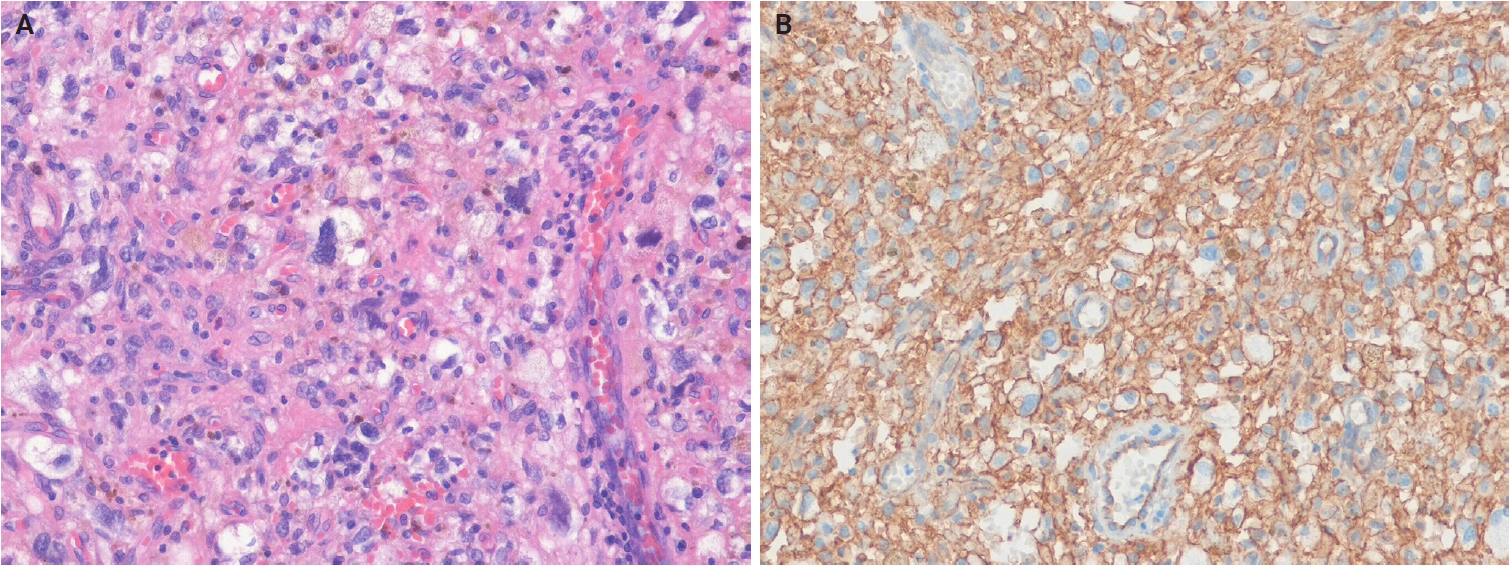
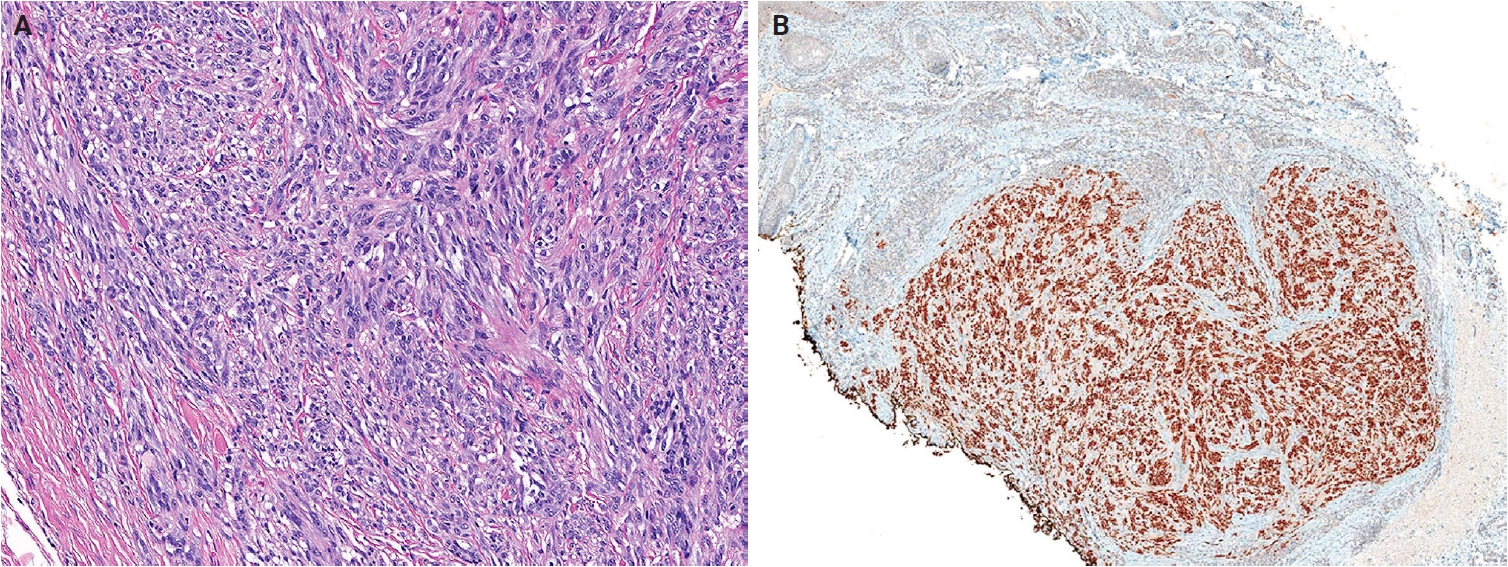

















Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
Fig. 6.
Fig. 7.
Fig. 8.
Fig. 9.
Fig. 10.
Fig. 11.
Fig. 12.
Fig. 13.
Fig. 14.
Fig. 15.
Fig. 16.
Fig. 17.
| Adipocytic tumors | Other benign |
| Benign | Lymphangioma (superficial lymphatic malformation) |
| Naevus lipomatosus superficialis | Cutaneous epithelioid angiomatous nodule |
| Lipoma | Postradiation atypical vascular lesion |
| Angiolipoma | Intermediate |
| Spindle cell/pleomorphic lipoma | Pseudomyogenic hemangioendothelioma |
| Intermediate | Epithelioid hemangioendothelioma |
| Atypical lipomatous tumor | Hobnail hemangioendotheliomas |
| Malignant | Composite hemangioendothelioma |
| Pleomorphic liposarcoma | Kaposi sarcoma |
| Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Malignant |
| Benign | Cutaneous angiosarcoma |
| Fibroma of tendon sheath | Pericytic and perivascular tumors |
| Calcifying aponeurotic fibroma | Benign |
| Sclerotic fibroma | Glomus tumor |
| Nuchal-type fibroma | Myopericytoma |
| Gardner fibroma | Myofibroma and myofibromatosis |
| Pleomorphic fibroma | Angioleiomyoma |
| Elastofibroma | Smooth muscle tumors |
| Desmoplastic fibroblastoma | Benign |
| Fibrous papule | Smooth muscle hamartoma |
| Fibroblastic connective tissue nevus | Cutaneous leiomyomas |
| Fibro-osseous tumor of digits | EBV-associated smooth muscle tumor |
| Dermatofibroma (fibrous histiocytoma) | Intermediate |
| Superficial fibromatosis | Atypical intradermal smooth muscle neoplasm |
| Inclusion body fibromatosis | Neural tumors |
| Plexiform fibrohistiocytic tumor | Benign |
| Superficial acral fibromyxoma | Dermal hyperneury/epithelial sheath neuroma |
| Cutaneous myxoma (superficial angiomyxoma) | Solitary circumscribed neuroma |
| Dermatomyofibroma | Dermal nerve sheath myxoma |
| Multinucleate cell angiohistiocytoma | Perineurioma |
| Plaque-like CD34-positive dermal fibroma | Neurofibroma |
| Nodular fasciitis | Schwannoma |
| EWSR1::SMAD3-rearranged fibroblastic tumor | Granular cell tumor |
| Intermediate | Hybrid nerve sheath tumors |
| Dermatofibrosarcoma protuberans | Malignant |
| Myxoinflammatory fibroblastic sarcoma | Malignant peripheral nerve sheath tumor |
| Malignant | Tumors of uncertain differentiation |
| Myxofibrosarcoma | Benign |
| Vascular tumors | Cellular neurothekeoma |
| Hemangioma | Epithelioid fibrous histiocytoma |
| Cherry hemangioma | Non-neural granular cell tumor |
| Sinusoidal hemangioma | PEComa |
| Microvenular hemangioma | Intermediate |
| Hobnail hemangioma | Angiomatoid fibrous histiocytoma |
| Glomeruloid hemangioma | NTRK-rearranged spindle cell neoplasm |
| Papillary hemangioma | Atypical fibroxanthoma |
| Spindle cell hemangioma | Superficial CD34-positive fibroblastic tumor |
| Epithelioid hemangioma | Malignant |
| Tufted hemangioma | Pleomorphic dermal sarcoma |
| Angiokeratoma | Epithelioid sarcoma |
| Infantile hemangioma | CRTC1::TRIM11 cutaneous tumor |
| Congenital non-progressive hemangiomas: Rapidly involuting congenital hemangioma and non-involuting congenital hemangioma | Dermal clear cell sarcoma |
| Lobular capillary hemangioma | Ewing sarcoma |
| Poikilodermatous plaque-like hemangioma | |
| Acquired elastotic hemangioma | |
| Verrucous venous malformation | |
| Arteriovenous malformation |
| Tumor family | Biological potential | Tumor type | Clinical features | Histological features | Immunohistochemistry | Molecular features |
|---|---|---|---|---|---|---|
| Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Benign | Fibrous papule | Middle-aged adults; sporadic in most cases; multiple early childhood lesions in tuberous sclerosis; nose, central face | Dome-shaped lesion, with dilated dermal blood vessels; spindle-shaped or stellate fibroblasts; fibrotic or collagenized stroma | CD34 (+); factor XIIIa (+) | Germline mutations in TSC1 or TSC2 (tuberous sclerosis-associated cases) |
| Benign | Fibroblastic connective tissue nevus | Young age (median, 10 years); slight female predominance; trunk, head and neck, limbs | Bland spindle-shaped and ovoid cells arranged in short, intersecting fascicles; entrapped and preserved skin appendages | CD34 (+); SMA (+) | KHDRBS1::NTRK3 (1 case) | |
| Benign | Fibro-osseous tumor of digit | Young adults; rapidly growing; subcutaneous tissues of the proximal phalanx (fingers and toes) | Spindle cells are arranged randomly or in short intersecting fascicles; unmineralized woven bone; vascular or myxoid stroma | SMA (+) in myofibroblasts; SATB2 (+) in osteoblast | USP6 rearrangement | |
| Benign | Inclusion body fibromatosis | Most cases <3 years; ~30% present at birth; dorsal/dorsolateral distal or middle phalanges of 2nd–4th digits (hands and feet) | Spindled cells with bland nuclei and light eosinophilic cytoplasm; a rounded, pale pink, intracytoplasmic inclusion | SMA (+); calponin (+); desmin (+); inclusions: calponin-1 (+), caldesmon (+) | - | |
| Benign | Multinucleate cell angiohistiocytoma | Middle-aged adults; solitary or multiple; dorsal hands, fingers, lower extremities, trunk, back, rarely head and neck | Triad of multinucleated and angulated stromal cells, dermal fibrosis parallel to the epidermis, and increased small-caliber vessels | CD68 (±); factor XIIIa (+) | - | |
| Benign | EWSR1::SMAD3-rearranged fibroblastic tumor | Wide age range (1–68 years; median, 39 years); female predominance; acral sites, most commonly the hands and feet | Hyalinized acellular center with peripheral zones composed of intersecting fascicles of fibroblastic spindle cells; calcification | ERG (+); CD34 (–); SMA (–) | EWSR1::SMAD3 | |
| Vascular tumors | Benign | Papillary hemangioma | Adult men; exclusively head and neck | Dilated, thin-walled dermal vascular channels with numerous luminal papillary projections; intracytoplasmic eosinophilic globules | CD31 (+); CD34 (+); ERG (+) | - |
| Benign | Poikilodermatous plaque-like hemangioma | Elderly men; lower extremities; usually solitary, rarely multiple plaques | Band-like proliferation of small vessels in the superficial dermis; no grenz zone; loss or reduction of elastic fibers | CD31 (+); CD34 (+); ERG (+) | - | |
| Benign | Acquired elastotic hemangioma | Middle-aged to elderly adults; slight female predominance; forearm, lateral neck; associated with chronic sun damage | Band-like proliferation of capillaries surrounded by pericytes; arranged parallel to the reticular dermis, in a background of solar elastosis | CD31 (+); CD34 (+); ERG (+) | - | |
| Intermediate | Hobnail hemangioendothelioma | PILA: infant, proximal extremities, buttock, or thigh; RHE: children or young adults, distal extremities | Hobnail endothelial cells, PILA: poorly defined slit-like or dilated lymphatic channels; RHE: infiltrative, branching vascular channels | Endothelial markers (CD31, ERG, FLI1) (+); lymphatic endothelial markers (podoplanin [D2-40], VEGFR3, PROX1) (+); loss of YAP1 C-terminus expression |
PILA: unknown; RHE: YAP1 rearrangements, most commonly YAP1::MAML2 (subset) | |
| Pericytic and perivascular tumors | Benign | Myofibroma and myofibromatosis | Most cases are in children; sporadic or familial; skin or subcutaneous tissue of the head and neck, upper extremities, trunk | Biphasic pattern of small primitive rounded cells and plump spindled myoid cells; hemangiopericytoma-like vessels | SMA (+); caldesmon (±); desmin (–) | Germline PDGFRB and NOTCH3 mutations (familial); somatic PDGFRB mutations (sporadic); SRF rearrangement (cellular cases) |
| Smooth muscle tumors | Benign | Smooth muscle hamartoma | Broad age spectrum; usually solitary; rare multiple or familial; trunk, extremities, head and neck, external genitalia | Haphazardly arranged bundles of mature smooth muscle in the dermis, often near pilosebaceous units | SMA (+); desmin (+); caldesmon (+) | ACTB mutations (subset) |
| Intermediate | EBV-associated smooth muscle tumor | Wide age distribution; immunosuppressed patients; CNS, liver, lung, subcutaneous tissues of the limbs and trunk | Interlacing fascicles of spindled myoid cells; admixture of smaller rounded cells; intratumoral lymphocytes | SMA (+); desmin (+); caldesmon (+); EBER in situ hybridization (+) | MYC overexpression (subset) | |
| Neural tumors | Benign | Dermal hyperneury/epithelial sheath neuroma | Syndromic cases in children; sporadic cases usually present in older adults; female predominance; lips, eyelid, tongue, trunk | Dermal hyperneury: enlarged, prominent dermal neural fibers; epithelial sheath neuroma: enlarged nerve fibers ensheathed by squamous epithelium | S100 protein (+); neurofilament (+); CD57 (+) in nerve fibers; CKs in perineural epithelial sheaths | - |
| Benign | Hybrid nerve sheath tumors | Any age (peak in young adults); wide anatomic sites; HSP is sporadic; HSN is associated with NF1 and NF2; HNP is associated with NF1 | HSP: admixture of Schwann and perineurial cells; HSN: schwannomatous nodules or Schwann cell bundles within neurofibroma; HNP: plexiform neurofibroma with areas of perineurial differentiation | S100 protein (+) and SOX10 (+) in Schwann cells; EMA (+), claudin-1 (+), and GLUT1 (+) in perineurial cells; CD34 (+) in fibroblasts | VGLL3 rearrangements (most HSPs) | |
| Tumors of uncertain differentiation | Benign | Epithelioid fibrous histiocytoma | Young to middle-aged adults; female predominance; extremities, trunk, head and neck | Exophytic lesions with adnexal collarette; uniform epithelioid cells with abundant eosinophilic to amphophilic cytoplasm | ALK (+) (~90%); EMA (+) (65%); CD30 (±) | ALK rearrangements (~90%); PRKC rearrangements (rare) |
| Benign | PEComa | Wide age (15–81 years); peak incidence in middle age; female predominance; extremities (lower limbs), trunk, head and neck | Epithelioid cells with granular, eosinophilic to clear cytoplasm; nested or trabecular architecture; perivascular growth; rare malignant features (mitotic activity, necrosis, and pleomorphism) | Melanocytic markers (HMB-45, melan-A [MART1], MITF) (+); smooth muscle markers (SMA, desmin, caldesmon) (+) | Frequent deletion of 16p; loss of heterozygosity at TSC2; TP53 mutations (63% of TSC2-mutated tumors) | |
| Intermediate | Angiomatoid fibrous histiocytoma | Wide age range; peak in the first two decades of life; extremities, trunk, head and neck | Fascicles or sheets of ovoid and spindled cells; pseudoangiomatoid spaces; peripheral cuff of lymphoplasmacytic cells | SOX9 (+); desmin (+) (~50%); EMA (±); CD99 (±); CD68 (±) | EWSR1::CREB1 (most frequent); EWSR1::ATF1, FUS::ATF1 (rare) | |
| Intermediate | NTRK-rearranged spindle cell neoplasm | Any age, most commonly pediatric; no sex predilection; extremities, trunk, head and neck | Haphazardly arranged spindled cells; infiltrative growth within fat resembling lipofibromatosis; distinctive stromal and perivascular keloidal collagen | CD34 (+); S100 protein (+); pan-TRK (+); SMA (±); SOX10 (–) | NTRK1 rearrangements: LMNA::NTRK1, TPR::NTRK1, or TPM3::NTRK1; others: NTRK2, NTRK3 rearrangements | |
| Intermediate | Superficial CD34-positive fibroblastic tumor | Middle-aged adults; slight male predominance; lower extremities (especially thigh), arm, buttock, shoulder, vulva | Spindle and pleomorphic cells with abundant, granular to glassy cytoplasm; marked nuclear pleomorphism; low mitotic activity | CD34 (+); PRDM10 (+); CADM3 (SynCAM3) (+); CKs (+) (~70%) | PRDM10 rearrangement (~60%) | |
| Malignant | CRTC1::TRIM11 cutaneous tumor | Broad age range (median, 44 years); extremities, trunk, head; oral and nasal mucosa (rare); slow-growing, skin-colored nodule; generally no pigment | Dermal-based, circumscribed; epithelioid to fusiform cells with vesicular nuclei, prominent nucleoli, and pale cytoplasm; intersecting fascicles; | Melanocytic markers (SOX10, S100 protein, melan-A [MART1]) (+) |
CRTC1::TRIM11 |
| Tumor category | Tumor type | Molecular genetic alterations | Immunohistochemistry |
|---|---|---|---|
| Adipocytic tumors | Naevus lipomatosus superficialis | Deletion of 2p24 (1 case) | - |
| Lipoma | Chromosome 12q13-15 rearrangements involving HMGA2; chromosome 16p21 rearrangements involving HMGA1 | HMGA2 (+) | |
| Angiolipoma | Mutations in PRKD2 and PIK3CA | - | |
| Spindle cell/pleomorphic lipoma | Partial or complete loss of chromosome 13q14 (RB1 locus) and/or 16q deletions | Loss of RB1 expression; CD34 (+) | |
| Atypical lipomatous tumor | Amplification of MDM2, CDK4, TSPAN31 (SAS), HMGA2, and CPM at 12q13-q15 region | MDM2 (+); CDK4 (+) | |
| Pleomorphic liposarcoma | Frequent loss of TP53, RB1, and NF1; frequent gain in 1p21, 1q21-q22, 5p13-p15, and 7q22 | - | |
| Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Fibroma of tendon sheath | USP6 fusion with various partners (PKM, RCC1, ASPN, COL1A1, COL3A1, MYH9, MIR22HG, CTNNB1, SPARC, CAP1, EMP1, CYTOR, NR1D1, RAB1A, and TNC) | - |
| Calcifying aponeurotic fibroma | FN1::EGF | EGF overexpression | |
| Sclerotic fibroma | Loss of PTEN (Cowden syndrome) | CD34 (+); loss of PTEN expression (Cowden syndrome) | |
| Nuchal-type fibroma | APC mutations (a few cases); MUTYH mutation (1 case) | May show β-catenin nuclear expression | |
| Gardner fibroma | Germline APC mutations (FAP-associated tumors) | CD34 (+); β-catenin nuclear expression (FAP-associated tumors) | |
| Pleomorphic fibroma | RB1 deletion; germline mutations in TP53 (Li-Fraumeni syndrome) | Loss of RB1 expression; CD34 (+) | |
| Elastofibroma | Copy gains (Xq); alterations to chromosome 1; t(8;12)(q22;q24.3); t(2;19) and t(X;1) rearrangements (some cases) | - | |
| Desmoplastic fibroblastoma | t(2;11)(q31;q12); t(11;17)(q12;p11.2) | FOSL1 overexpression | |
| Dermatofibroma (fibrous histiocytoma) | Recurrent gene fusions involving PKC isoforms (PRKCA, PRKCB, and PRKCD) with various membrane-associated partners (LAMTOR1, PDPN, CD63, and KIRREL family genes) (minority of cases) | SMA (+); desmin (+) (~30%); CD34 (+) (5%) | |
| Superficial fibromatosis | Lack of CTNNB1 and APC mutations | β-catenin nuclear expression (rare cases); SMA (±); desmin (±) | |
| Plexiform fibrohistiocytic tumor | 46,X,del(X)(q13)[3]/46,XX[23], 46,XY,t(4;15)(q21;q15), and 46,XY,-6,-8,del(4)(q25q31),del(20)(q11.2) with additional der (8)(p22) (1 case) | SMA (+) in myofibroblasts; CD68 (+) in histiocytes | |
| Superficial acral fibromyxoma | RB1 deletions | Loss of RB1 expression; CD34 (+); CD99 (+); nestin (+) | |
| Cutaneous myxoma (superficial angiomyxoma) | PRKAR1A mutations (45%−65% of Carney complex); MYH8 mutations | Loss of PRKAR1A expression; SMA (+) | |
| Nodular fasciitis | USP6 rearrangements (~90%); MYH9::USP6 (>65%); PPP6R3::USP6 and CALD1::USP6 (malignant cases); EIF5A::USP6 (dermal nodular fasciitis with atypical mitosis) | SMA (+) | |
| Dermatofibrosarcoma protuberans | COL1A1::PDGFB (>90%); PDGFD rearrangements with COL6A3, EMILIN2, or TNC (<5%) | CD34 (+) | |
| Myxoinflammatory fibroblastic sarcoma | VGLL3 amplification (>50%); TGFBR3::MGEA5 (13%); BRAF fusions or amplifications | Cyclin D1 (+); factor XIIIa (+); podoplanin (D2-40) (+); CD10 (+); CD34 (±); CD163 (±); PRAME (±); CKs (±); CD68 (±); SMA (±) | |
| Myxofibrosarcoma | Frequent 5p gains with coamplification of TRIO, RICTOR, SKP2, and AMACR (associated with higher grade); inactivating mutations in TP53 (46%), RB1 (18%), and/or CDKN2A/2B (16%) | SMA (±); CD34 (±) | |
| Vascular tumors | Cherry hemangioma | Frequent somatic mutations of GNA14, GNA11, and GNAQ; rare somatic mutations in HRAS and KRAS | - |
| Spindle cell hemangioma | IDH1 p.R132C hotspot mutations (sporadic lesions and Maffucci syndrome); frequent somatic alterations in 2p22.3, 2q24.3, and 14q11.2 (Maffucci syndrome) | Endothelial markers (CD31, CD34, ERG) (+) in endothelial cells; SMA (±) and/or desmin (±) in spindle cells | |
| Epithelioid hemangioma | FOS or FOSB gene rearrangements | Endothelial markers (CD31, CD34, ERG) (+) in endothelial cells; FOSB (+) (FOSB-rearranged cases); SMA-positive pericytes | |
| Infantile hemangioma | No identified genetic abnormalities | GLUT1 (+) | |
| Congenital non-progressive hemangioma: RICH and NICH | Somatic activating mutations in GNAQ and GNA11 (RICH and NICH) | GLUT1 (–) | |
| Lobular capillary hemangioma | RAS-family and BRAF mutations (mainly c.1799T>A p.V600E) | - | |
| Verrucous venous malformation | Somatic missense mutation in MAP3K3 (some cases) | GLUT1 (±) |
|
| Arteriovenous malformation | Germline RASA1 mutations (hereditary CM-AVM syndrome) | GLUT1 (–) | |
| Lymphangioma (superficial lymphatic malformation) | Mutations in FLT4 (families with Milroy disease); mutations in PROX1 and FOXC2 | Lymphatic endothelial markers (podoplanin [D2-40], LYVE1, PROX1) (+); endothelial markers (CD31, ERG) (+) | |
| Postradiation atypical vascular lesion | Mutations in HRAS and TERT (rare cases) | Endothelial markers (CD31, CD34, ERG) (+); lymphatic endothelial markers (podoplanin [D2-40], LYVE1, PROX1) (+) | |
| Pseudomyogenic hemangioendothelioma | FOSB rearrangement, including SERPINE1::FOSB, ACTB::FOSB, WWTR1::FOSB, CLTC::FOSB, and EGFL7::FOSB (more aggressive) | CKs (+); FLI1 (+); ERG (+); FOSB (+); CD31 (+) (~50%); SMA (+) (~30%); CD34 (–) | |
| Epithelioid hemangioendothelioma | WWTR1::CAMTA1 (>90%); YAP1::TFE3 (rare subtype) | CAMTA1 (+) in EHE with WWTR1::CAMTA1 fusion; TFE3 (+) and loss of YAP1 C-terminus expression in EHE with YAP1::TFE3 fusion | |
| Composite hemangioendothelioma | YAP1::MAML2 (~27%); PTBP1::MAML2 (3 cases of neuroendocrine CHE); EPC1::PCH2 (1 case of neuroendocrine CHE) | Loss of YAP1 C-terminus expression in CHE with YAP1::MAML2 fusions; synaptophysin (+) in neuroendocrine CHE | |
| Kaposi sarcoma | Gain at 11q13 with FGF3 and FGF4 amplification; Y chromosome loss (early lesions); copy-number changes involving chromosomes 16, 17, 21, X, and Y (late lesions) | KSHV/HHV8-associated protein LANA-1 (+); CD31 (+); CD34 (+); ERG (+); podoplanin (D2-40) (+) | |
| Cutaneous angiosarcoma | Mutations in KDR, PTPRB, and PLCG1 (~40%); MYC amplification in >90% of radiation- and lymphedema-associated angiosarcoma;CIC alterations (younger patients) | MYC (+) |
|
| Pericytic and perivascular tumors | Glomus tumor | Mutations in GLMN (familial); MIR143 and NOTCH genes (NOTCH1, NOTCH2, or NOTCH3) rearrangements (sporadic); BRAF p.V600E mutations (rare malignant tumors) | SMA (+); caldesmon (+); collagen IV (+); RGS5 (+) |
| Myopericytoma | BRAF mutations (15%) | SMA (+); caldesmon (+); MSA (+) | |
| Angioleiomyoma | Monosomy of chromosome 13; loss of 6p, 21q, and 13q; NOTCH gene fusions (very small subset) | SMA (+); calponin (+); caldesmon (+); desmin (±); RGS5 (+) | |
| Smooth muscle tumors | Cutaneous leiomyomas | Germline mutations in FH (HLRCC-associated tumors) | SMA (+); desmin (+); caldesmon (+); FH (–) and 2SC (+) in HLRCC-associated tumors |
| Neural tumors | Perineurioma | Deletion of 22q12, monosomies, and mutations in NF2; deletion of 17q11 (soft tissue subtype); 10q24 rearrangement (sclerosing subtype); loss of 13q and small deletions of chromosomes 3, 6, and 9 (malignant perineurioma) | EMA (+); collagen IV (+); laminin (+); claudin-1 (+); GLUT1 (+); CD34 (+) (~60%) |
| Neurofibroma | Mutations in NF1 gene (multiple or plexiform and diffuse subtypes); loss of CDKN2A (ANNUBP) | S100 protein (+); SOX 10 (+); CD34 (+) in perineurial fibroblasts; peripherin (+) in thin axons | |
| Schwannoma | NF2 mutations with complete or partial loss of chromosome 22; alterations in ARID1A/ARID1B (~29%), TSC1/TSC2 (~15%), or SH3PXD2A::HTRA1 (~10%); germline mutations in NF2, LZTR1, SMARCB1, and DGCR8 (associated with schwannoma susceptibility) | S100 protein (+); SOX10 (+); EMA (+) in perineurial capsule | |
| Granular cell tumor | Loss-of-function mutations in ATP6AP1 and ATP6AP2 (~72%) | S100 protein (+); SOX10 (+); CD68 (+); inhibin (±); calretinin (±); TFE3 (±); MITF (±) | |
| Malignant peripheral nerve sheath tumor | Mutations in NF1, CDKN2A/CDKN2B, and PRC2 core components (EED or SUZ12); complete loss of PRC2 activity and H3K27me3 expression (~80% of high-grade MPNSTs); SMARCB1 inactivation (~75% of epithelioid subtype) | S100 protein (+) (<50%); SOX10 (+) (<70%); GFAP (+) (<20%–30%); loss of H3K27me3 expression |
|
| Tumors of uncertain differentiation | Non-neural granular cell tumor | ALK rearrangements (60%) | CD68 (+); CD63 (NKI/C3) (+); ALK (+) (subset) |
| Atypical fibroxanthoma | UV-radiation signature mutations in TP53; 9p and 13q deletions; TERT mutations | CD10 (+); CD68 (±); CD163 (±) | |
| Pleomorphic dermal sarcoma | Mutations in TP53, NOTCH family genes, and TERT promoter; alterations in CDKN2A and CDKN2B | CD10 (+); CD99 (+); PDGFRB (±); SMA (±); CD31 (±) | |
| Epithelioid sarcoma | Monoallelic or biallelic SMARCB1 deletion | Loss of SMARCB1 (INI1); CKs (+); EMA (+); CD34 (+) (>50%); ERG (+) (40%−67%) | |
| Dermal clear cell sarcoma | EWSR1::ATF1 (70%−90%); EWSR1::CREB1 (~5%) | S100 protein (+); SOX10 (+); PMEL (gp100) (+); TYRP1 (gp75) (+); melan-A (MART1) (+); MITF (+); PRAME (±) |
|
| Ewing sarcoma | EWSR1::FLI1 (~85%); EWSR1::ERG (5%−10%); EWSR1::FEV (rare); EWSR1::ETV1 (rare); FUS::ERG (rare) | CD99 (+); NKX2.2 (+); FLI1 (+) and ERG (+) |
| Tumor family | 4th edition WHO classification of skin tumors and 5th edition WHO classification of soft tissue tumors | 5th edition WHO classification of skin tumors |
|---|---|---|
| Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Collagenous fibroma | Desmoplastic fibroblastoma |
| Acral fibromyxoma | Superficial acral fibromyxoma | |
| EWSR1-SMAD3–positive fibroblastic tumor (emerging) | EWSR1::SMAD3-rearranged fibroblastic tumor | |
| Vascular tumors | Atypical vascular lesion | Postradiation atypical vascular lesion |
| Retiform hemangioendothelioma | Hobnail hemangioendothelioma | |
| Smooth muscle tumors | Cutaneous leiomyosarcoma (atypical smooth muscle tumor) | Atypical intradermal smooth muscle neoplasm |
| Tumors of uncertain differentiation | Primitive non-neural granular cell tumor | Non-neural granular cell tumor |
| NTRK-rearranged spindle cell neoplasm (emerging) | NTRK-rearranged spindle cell neoplasm |
| Tumor type | 4th WHO classification of skin tumors and 5th WHO classification of soft tissue tumors | 5th WHO classification of skin tumors |
|---|---|---|
| Epithelioid fibrous histiocytoma | Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Tumors of uncertain differentiation |
| Myofibroma and myofibromatosis | Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Pericytic and perivascular tumors |
| Superficial CD34-positive fibroblastic tumor | Fibroblastic, myofibroblastic, and fibrohistiocytic tumors | Tumors of uncertain differentiation |
| Myxofibrosarcoma | Tumors of uncertain differentiation | Fibroblastic, myofibroblastic, and fibrohistiocytic tumors |
WHO, World Health Organization; EBV, Epstein-Barr virus; PEComa, perivascular epithelioid cell tumor; NTRK, neurotrophic tyrosine receptor kinase.
WHO, World Health Organization; SMA, smooth muscle actin; SATB2, special AT-rich sequence-binding protein 2; ERG, ETS-related gene; PILA, papillary intralymphatic angioendothelioma; RHE, retiform hemangioendothelioma; FLI1, Friend leukemia virus integration 1; VEGFR3, vascular endothelial growth factor receptor 3; PROX1, prospero homeobox 1; YAP1 C-terminus, yes-associated protein 1 C-terminus; EBV, Epstein-Barr virus; CNS, central nervous system; EBER, Epstein-Barr virus-encoded small RNAs; CK, cytokeratin; HSP, hybrid schwannoma/perineurioma; HSN, hybrid schwannoma/neurofibroma; NF1, neurofibromatosis type 1; NF2, neurofibromatosis type 2; HNP, hybrid neurofibroma/perineurioma; SOX10, SRY-box transcription factor 10; EMA, epithelial membrane antigen; GLUT1, glucose transporter 1; ALK, anaplastic lymphoma kinase; PEComa, perivascular epithelioid cell tumor; HMB-45, human melanoma black 45; MART1, melanoma antigen recognized by T cells 1; MITF, microphthalmia-associated transcription factor; SOX9, SRY-box transcription factor 9; NTRK, neurotrophic tyrosine receptor kinase; pan-TRK, pan–tropomyosin receptor kinase; PRDM10, PR domain–containing protein 10; CADM3, cell adhesion molecule 3; SynCAM3, synaptic cell adhesion molecule 3; TRIM11, tripartite motif–containing 11; +, positive staining; ±, variable staining; –, negative staining. Loss of expression of YAP1 C-terminus is seen in subsets of RHE with Limited synaptophysin expression may be seen in PILA and RHE; All tumors show diffuse SOX10 expression, whereas diffuse S100 protein staining is present in only about 50% of cases and melan-A (MART1) and HMB-45 are expressed only focally.
HMGA2, high mobility group AT-hook 2; RB1, retinoblastoma 1; MDM2, mouse double minute 2; CDK4, cyclin-dependent kinase 4; EGF, epidermal growth factor; PTEN, phosphatase and tensin homolog; FAP, familial adenomatous polyposis; FOSL1, FOS-like antigen 1; SMA, smooth muscle actin; PRKAR1A, protein kinase cAMP-dependent regulatory subunit type I alpha; PRAME, preferentially expressed antigen in melanoma; CK, cytokeratin; ERG, ETS-related gene; FOSB, FBJ murine osteosarcoma viral oncogene homolog B; GLUT1, glucose transporter 1; RICH, rapidly involuting congenital hemangioma; NICH, non-involuting congenital hemangioma; CM-AVM, capillary malformation–arteriovenous malformation; LYVE1, lymphatic vessel endothelial hyaluronan receptor 1; PROX1, prospero homeobox 1; FLI1, Friend leukemia virus integration 1; CAMTA1, calmodulin-binding transcription activator 1; EHE, epithelioid hemangioendothelioma; TFE3, transcription factor E3; YAP1 C-terminus, yes-associated protein 1 C-terminus; CHE, composite hemangioendothelioma; KSHV, Kaposi sarcoma-associated herpesvirus; HHV8, human herpesvirus 8; LANA-1, latency-associated nuclear antigen-1; MYC, MYC proto-oncogene; H3K27me3, histone H3 lysine 27 trimethylation; EMA, epithelial membrane antigen; RGS5, regulator of G-protein signaling 5; MSA, muscle-specific actin; HLRCC, hereditary leiomyomatosis and renal cell carcinoma; FH, fumarate hydratase; 2SC, 2-succinylcysteine; ANNUBP, atypical neurofibromatous neoplasm of uncertain biological potential; SOX10, SRY-box transcription factor 10; MITF, microphthalmia-associated transcription factor; PRC2, polycomb repressive complex 2; GFAP, glial fibrillary acidic protein; ALK, anaplastic lymphoma kinase; NOTCH, neurogenic locus notch homolog; PDGFRB, platelet-derived growth factor receptor beta; SMARCB1, SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily B, member 1; INI1, integrase interactor 1; PMEL, premelanosome protein; TYRP1, tyrosinase-related protein 1; MART1, melanoma antigen recognized by T cells 1; NKX2.2, NK2 homeobox 2; +, positive staining; ±, variable staining; –, negative staining. Focal and weak endothelial GLUT1 positivity may be seen; Radiation- and lymphedema-associated angiosarcomas are almost always positive for MYC but may occasionally be negative; Complete loss of H3K27me3 expression occurs more frequently in high-grade and radiation-induced MPNSTs than in low-grade MPNSTs; PRAME expression is usually absent or only focal; FLI1 and ERG are often expressed in cases with the corresponding translocation.
WHO, World Health Organization; NTRK, neurotrophic tyrosine receptor kinase.
WHO, World Health Organization.

