 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 49(3); 2015 > Article
-
Original Article
IDH Mutation Analysis in Ewing Sarcoma Family Tumors - Ki Yong Na, Byeong-Joo Noh1, Ji-Youn Sung2, Youn Wha Kim2, Eduardo Santini Araujo3, Yong-Koo Park2
-
Journal of Pathology and Translational Medicine 2015;49(3):257-261.
DOI: https://doi.org/10.4132/jptm.2015.04.14
Published online: May 15, 2015
Department of Pathology, Central Physical Examination Agency, Daegu, Korea
1Department of Pathology, Graduate School of Medicine, Kyung Hee University, Seoul, Korea
2Department of Pathology, Kyung Hee University College of Medicine, Seoul, Korea
3Orthopedic Pathology Laboratory, Buenos Aires, Argentina
- Corresponding Author Yong-Koo Park, M.D. Department of Pathology, Kyung Hee University Hospital, 23 Kyungheedae-ro, Dongdaemun-gu, Seoul 130-872, Korea Tel: +82-2-958-8742 Fax: +82-2-958-8740 E-mail: ykpark@khmc.or.kr
© 2015 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background:
- Isocitrate dehydrogenase (IDH) catalyzes the oxidative decarboxylation of isocitrate to yield α-ketoglutarate (α-KG) with production of reduced nicotinamide adenine dinucleotide (NADH). Dysfunctional IDH leads to reduced production of α-KG and NADH and increased production of 2-hydroxyglutarate, an oncometabolite. This results in increased oxidative damage and stabilization of hypoxia-inducible factor α, causing cells to be prone to tumorigenesis.
-
Methods:
- This study investigated IDH mutations in 61 Ewing sarcoma family tumors (ESFTs), using a pentose nucleic acid clamping method and direct sequencing.
-
Results:
- We identified four cases of ESFTs harboring IDH mutations. The number of IDH1 and IDH2 mutations was equal and the subtype of IDH mutations was variable. Clinicopathologic analysis according to IDH mutation status did not reveal significant results.
-
Conclusions:
- This study is the first to report IDH mutations in ESFTs. The results indicate that ESFTs can harbor IDH mutations in previously known hot-spot regions, although their incidence is rare. Further validation with a larger case-based study would establish more reliable and significant data on prevalence rate and the biological significance of IDH mutations in ESFTs.
- Patient and tissue samples
- Formalin-fixed, paraffin-embedded tissue samples from 61 patients with primary localized ESFTs were obtained in Korea, Brazil, and Argentina. Fifty-five of the 61 tissue samples were obtained by surgical biopsy and the other six were obtained by surgical excision. At the time of tissue sampling, none of the patients had a history of chemotherapy or radiation therapy and there was no evidence of metastatic disease. The disease was diagnosed according to World Health Organization (WHO) criteria [10]. Briefly, they are small round cell sarcomas showing diffuse membranous CD99 immunostaining, cytoplasmic periodic acid–Schiff staining, and EWSR1 gene translocation as demonstrated with fluorescence in situ hybridization (ZytoLight SPEC EWSR1 Dual Color Break Apart Probes, ZytoVision, Bremerhaven, Germany). However, lack of an EWSR1 gene translocation was not considered as grounds for exclusion if a tumor showed the typical immunophenotypes, which are inconsistent with other small round cell tumors in the differential diagnosis of diseases such as lymphoma and rhabdomyosarcoma.
- After histological diagnosis, the patients received standard multidrug chemotherapy in combination with surgery. Data, including the follow-up period and overall survival, were available for 48 patients. During follow-up, assessment of distant metastasis was available in 38 patients. The patients were grouped into dead of disease, alive with disease, and no evidence of disease (NED). The classification of patients as NED was established when the follow-up period had passed more than 24 months. Our study protocol was reviewed and approved by the Kyung Hee University Institutional Review Board.
- Pentose nucleic acid–mediated clamping polymerase chain reaction for detection of IDH1/2 mutations
- IDH1/2 mutations were tested using the pentose nucleic acid (PNA) Clamp IDH1/2 Mutation Detection Kit (Panagene, Daejeon, Korea). All reactions had a total reaction volume of 20 μL and contained template DNA, primer and PNA probe sets, and fluorescent polymerase chain reaction (PCR) master mix. All required reagents were included with the kit. Real-time PCR reactions of PNA-mediated clamping PCR were performed using a CFX 96 (Bio-Rad, Hercules, CA, USA). PCR cycling conditions were as follows: 5 minutes at 94ºC followed by 40 cycles of 94ºC for 30 seconds, 70ºC for 20 seconds, 63ºC for 30 seconds, and 72ºC for 30 seconds. In this assay, PNA probes and DNA primers were used together in the clamping reaction. Positive signals were detected by intercalation of fluorescent dye. The PNA probe, which is complementary to the wild-type sequence, suppresses amplification of the wild-type target. This suppression results in preferential amplification of mutant sequences by competitively inhibiting the binding of DNA primers to wild-type DNA. PCR efficiency was determined by measuring the threshold cycle (Ct) value. Ct values for control and mutant assays were obtained from fluorescent amplification plots. Calculations of the delta Ct (ΔCt) value were done as follows: ΔCt1=(Standard Ct)–(Sample Ct), ΔCt2=(Sample Ct)–(Non-PNA mix Ct). The gene was considered to be mutated when ΔCt1 values were more than 2.0. When ΔCt1 values were between 0 and 2, a ΔCt2 value was then calculated. The gene was considered to be mutated if the calculated ΔCt2 value was ≤4.
- Direct sequencing
- Genomic PCR for sequencing was performed in 20-μL volumes using 30 ng of template DNA and 2× Taq PCR Smart Mix (Solgent, Daejeon, Korea). The PCR primers used for IDH1/2 amplification were as follows: IDH1 forward primer (5'-CGGTCTTCAGAGAAGCCATT-3') and IDH1 reverse primer (5'-GCAAAATCACATTATTGCCAAC-3'). IDH2 forward primer (5'-CCAATGGAACTATCCG-3') and IDH2 reverse primer (5'-CTCCACCCTGGCCTACCTG-3'). PCR cycling commenced with a 10 minutes hold at 95ºC, followed by 40 cycles of 95ºC for 30 seconds, 58ºC for 40 seconds, and 72ºC for 60 seconds, terminating with 72ºC for 5 minutes. Each amplified product was purified using a PCR clean-up kit (Macherey-Nagel, Duren, Germany) and sequenced in duplicate, in both the forward and reverse directions, using a BigDye Terminator Kit (Applied Biosystems, Carlsbad, CA, USA) on an ABI Prism 3100 station (Applied Biosystems), according to the manufacturer’s instructions. Sequences were compared with the GenBank-archived sequence of human IDH1/2.
- Immunohistochemistry
- The primary antibody that is specific for the IDH1 R132H point mutation (1:200, Histonova DIA-H09, Dianova, Hamburg, Germany) was used for the samples bearing IDH1 mutations, revealed by either direct PCR or PNA clamping. The Bond Polymer Intense Detection System (Vision Biosystems, Melbourne, Australia) was used according to the manufacturer's instructions with minor modifications. Nuclei were counterstained with hematoxylin. Paraffin-embedded tissues of brain astrocytomas were used as a positive control.
- Statistics
- Statistical analyses were performed using SPSS Software (SPSS Inc., Chicago, IL, USA). Pearson’s chi-square test or Fisher exact test were performed to determine correlations between IDH mutation status and clinicopathological parameters. Statistical significance was defined as a p-value less than .05.
MATERIALS AND METHODS
- Using the PNA clamping method, IDH1/2 mutations were detected in three of the 61 patients (5%). Of these three samples, two were IDH1 mutants and one sample was an IDH2 mutant. By direct sequencing, IDH1/2 mutations were detected in two of the 61 patients (3%), of which, one sample was an IDH1 mutant and one sample was an IDH2 mutant. In total, four cases out of 61 (6%) harbored IDH1/2 mutations by at least one of the two methods employed, and the numbers of IDH1 and IDH2 mutants were equal (Table 1).
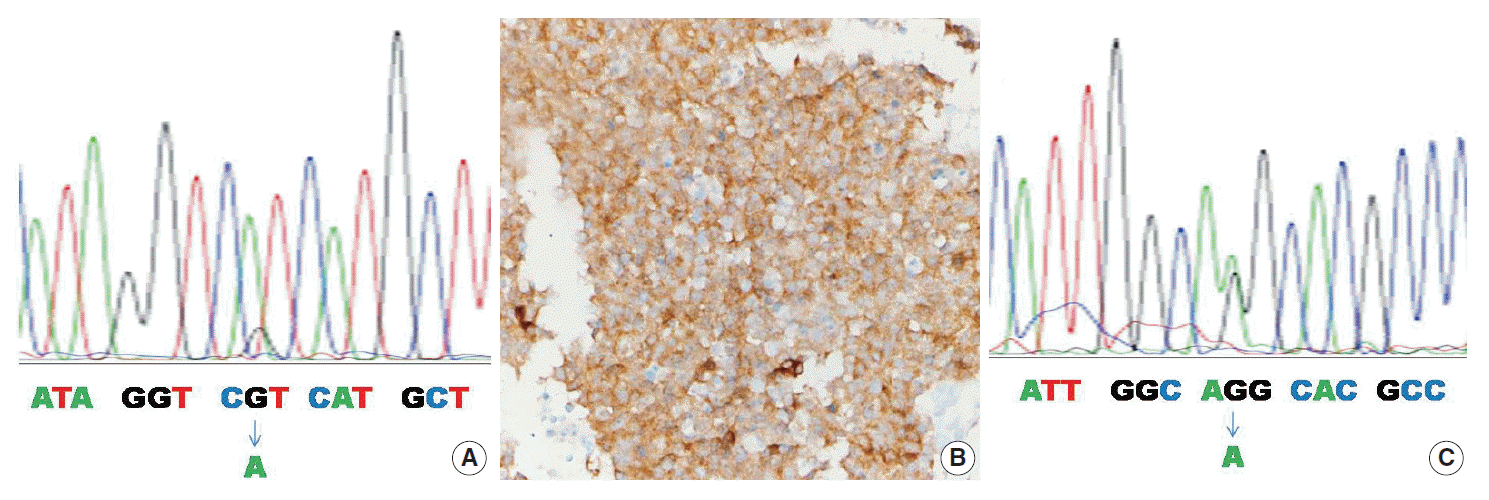
- Table 2 summarizes the clinicopathologic characteristics of the four mutant cases. In one of four cases, the IDH1 mutation was found by both the PNA clamping method and direct sequencing (case No. 1) (Fig. 1A). In two of four cases, the IDH1/2 mutation was found only by the PNA clamping method (cases Nos. 2 and 3). In one of four cases, examination by the PNA clamping method showed equivocal results, but direct sequencing showed an IDH2 mutation (case No. 4) (Fig. 1C). The overall concordance rate of both methods was over 95% (58 of 61) and the discordance rate was less than 5% (3 of 61). In mutant cases, the concordance rate was 25% (1 of 4) and the discordance rate was 75% (3 of 4), although case No. 4 showed equivocal results by the PNA clamping method. Immunohistochemistry with antibody to DIA-H09 in the four cases bearing IDH1/2 mutations showed positive reactions only in case No. 1 (Fig. 1B).
- In the four cases bearing IDH1/2 mutations, three patients were Korean and one patient was Brazilian, and the male/female ratio was 1:1. Three of the four patients were in their second decade and one patient was in the fifth decade. There was no evidence of distant metastasis in all patients and only one patient died during follow-up.
- Statistical analyses showed that IDH1/2 mutant cases had stronger associations with Korean patients than with South American patients (p=.009). There was no significant association between IDH1/2 mutations and any of the other characteristics of tumors or patients (Table 3).
RESULTS
- Research on IDH1/2 mutations in human tumors has been active in recent years and has revealed that various tumors of different origins bear IDH1/2 mutations [2]. Following increased interest in IDH1/2 mutations in soft tissue tumors, a rudimentary study on sarcoma cell lines demonstrated IDH mutations in fibrosarcoma [11]. Subsequently, a study on chondrogenic tumors demonstrated IDH1/2 mutations in 81 of 145 (56%) cases with an IDH1:IDH2 mutation ratio of 10.6:1. This study also included the evaluation of IDH1/2 mutations in 222 osteosarcomas, 79 chordomas, and 25 ESFTs, and no mutations were found [7]. Therefore, IDH mutations are considered to be found exclusively in chondrogenic tumors. Furthermore, previous studies support the value of examining IDH mutations for the purpose of differentiating chondrosarcoma from chondroblastic osteosarcoma [12] and chordoma [13]. However, a recent study in Japan showed that three of 12 osteosarcomas (25%) [8] and 16 of 20 giant cell tumors (80%) harbor IDH2 mutations [9], suggesting the possibility of IDH1/2 mutations in various soft tissue tumors in addition to chondrogenic tumors.
- We demonstrated that four of 61 ESFTs (6%) possessed IDH mutations. The PNA clamping method is known to be sensitive, rapid, and simple to perform and can detect mutant alleles when present at levels 100-fold lower than those of wild-type alleles. In contrast, the minimum percentage of mutant DNA required for analysis by direct sequencing is more than 25% [14]. The two cases harboring IDH mutations, found only by PNA clamping, might have had less than 25% mutant DNA. It is worth noting that three osteosarcomas bearing IDH mutations were found in Japanese [8], and three ESFT bearing IDH mutations were found in Korean patients. However, evaluation of the same tumors from American patients revealed no mutations [7]. Although it is still early to remark on the background responsible for these findings, it is possible that Asian populations may be predisposed to IDH mutations in these tumors and therefore should be further evaluated.
- Variation in the most prevalent mutation type according to tumor has been observed. IDH1 R132H represents the most common type in gliomas [4], and IDH2 R140Q is exclusively found in acute myeloid leukemia [5]. Whereas IDH1 R132C represents the most common type in cartilaginous tumors [13], IDH2 R172S is the dominant type in osteosarcomas [8] and giant cell tumors [9]. In our study, ESFTs demonstrated equal numbers of IDH1 and IDH2 mutations in which one case of R132H and one case of R172K were found. A previous study on cartilaginous tumors demonstrated that IDH mutations are frequent in acral-based tumors without any other association with other factors [7]. IDH mutations in osteosarcomas and giant cell tumors did not show any association with other clinical parameters [8,9]. Our study also did not find a significant association between IDH mutations and clinical parameters of ESFTs.
- In conclusion, our study is the first report to demonstrate IDH mutations in ESFTs. It provides evidence that ESFTs can harbor IDH mutations in previously known hot-spot regions, although its incidence is rare. To provide generalized knowledge, our study is still lacking enough data about these mutations in ESFTs. Further validation with a larger case-based study would establish more reliable and significant data on prevalence rate and the biological significance of IDH1/2 mutation status in ESFTs.
DISCUSSION
| PNA clamping | Direct sequencing | |
|---|---|---|
| Wild type | 57 | 59 |
| Mutant | ||
| IDH1 | 2 | 1 |
| IDH2 | 1 | 1 |
| Equivocal | 1 | 0 |
- 1. van Maldegem AM, Hogendoorn PC, Hassan AB. The clinical use of biomarkers as prognostic factors in Ewing sarcoma. Clin Sarcoma Res 2012; 2: 7.PubMedPMC
- 2. Schaap FG, French PJ, Bovée JV. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors. Adv Anat Pathol 2013; 20: 32-8. ArticlePubMed
- 3. Lu C, Venneti S, Akalin A, et al. Induction of sarcomas by mutant IDH2. Genes Dev 2013; 27: 1986-98. ArticlePubMedPMC
- 4. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765-73. PubMedPMC
- 5. Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 2010; 116: 2122-6. ArticlePubMedPDF
- 6. Abdel-Wahab O, Manshouri T, Patel J, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res 2010; 70: 447-52. ArticlePubMedPMCPDF
- 7. Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011; 224: 334-43. ArticlePubMedPDF
- 8. Liu X, Kato Y, Kaneko MK, et al. Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med 2013; 2: 803-14. ArticlePubMedPMCPDF
- 9. Kato Kaneko M, Liu X, Oki H, et al. Isocitrate dehydrogenase mutation is frequently observed in giant cell tumor of bone. Cancer Sci 2014; 105: 744-8. ArticlePubMedPMCPDF
- 10. Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press, 2013.
- 11. Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P, Gardner MB. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer 1974; 33: 1027-33. ArticlePubMed
- 12. Kerr DA, Lopez HU, Deshpande V, et al. Molecular distinction of chondrosarcoma from chondroblastic osteosarcoma through IDH1/2 mutations. Am J Surg Pathol 2013; 37: 787-95. ArticlePubMed
- 13. Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y. Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol 2012; 29: 201-6. ArticlePubMedPDF
- 14. Lee HJ, Xu X, Kim H, et al. Comparison of direct sequencing, PNA clamping-real time polymerase chain reaction, and pyrosequencing methods for the detection of EGFR mutations in non-small cell lung carcinoma and the correlation with clinical responses to EGFR tyrosine kinase inhibitor treatment. Korean J Pathol 2013; 47: 52-60. ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Targeting metastasis in paediatric bone sarcomas
Emma C. Bull, Archana Singh, Amy M. Harden, Kirsty Soanes, Hala Habash, Lisa Toracchio, Marianna Carrabotta, Christina Schreck, Karan M. Shah, Paulina Velasco Riestra, Margaux Chantoiseau, Maria Eugénia Marques Da Costa, Gaël Moquin-Beaudry, Pan Pantziark
Molecular Cancer.2025;[Epub] CrossRef - Targeting developmental vulnerabilities in childhood sarcomas
Elise Young, Barnaby Kelly, Jason E. Cain
Cancer and Metastasis Reviews.2025;[Epub] CrossRef - Ewing’s Sarcoma Presenting in the Paranasal Sinus – A Case Report
Yashika Kewalramani, Ajay Parihar, Prashanthi Reddy, Rashi Mandlik
Annals of Maxillofacial Surgery.2024; 14(2): 228. CrossRef - Glutamine-dependent effects of nitric oxide on cancer cells subjected to hypoxia-reoxygenation
Dianna Xing, Gloria A. Benavides, Michelle S. Johnson, Ran Tian, Stephen Barnes, Victor M. Darley-Usmar
Nitric Oxide.2023; 130: 22. CrossRef - Hypoxia and HIFs in Ewing sarcoma: new perspectives on a multi-facetted relationship
A. Katharina Ceranski, Martha J. Carreño-Gonzalez, Anna C. Ehlers, Maria Vittoria Colombo, Florencia Cidre-Aranaz, Thomas G. P. Grünewald
Molecular Cancer.2023;[Epub] CrossRef - Metabolic adaptations in cancers expressing isocitrate dehydrogenase mutations
Ingvild Comfort Hvinden, Tom Cadoux-Hudson, Christopher J. Schofield, James S.O. McCullagh
Cell Reports Medicine.2021; 2(12): 100469. CrossRef - Isocitrate dehydrogenase gene variants in cancer and their clinical significance
Thomas Cadoux-Hudson, Christopher J. Schofield, James S.O. McCullagh
Biochemical Society Transactions.2021; 49(6): 2561. CrossRef - Advances in sarcoma gene mutations and therapeutic targets
Peng Gao, Nicole A. Seebacher, Francis Hornicek, Zheng Guo, Zhenfeng Duan
Cancer Treatment Reviews.2018; 62: 98. CrossRef - Clinicopathologic Features of the Non-CNS Primary Ewing Sarcoma Family of Tumors in the Head and Neck Region
Chang Gok Woo, Bora Lee, Joon Seon Song, Kyung-Ja Cho
Applied Immunohistochemistry & Molecular Morphology.2018; 26(9): 632. CrossRef - EWS/FLI is a Master Regulator of Metabolic Reprogramming in Ewing Sarcoma
Jason M. Tanner, Claire Bensard, Peng Wei, Nathan M. Krah, John C. Schell, Jamie Gardiner, Joshua Schiffman, Stephen L. Lessnick, Jared Rutter
Molecular Cancer Research.2017; 15(11): 1517. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-


Fig. 1.
| PNA clamping | Direct sequencing | |
|---|---|---|
| Wild type | 57 | 59 |
| Mutant | ||
| IDH1 | 2 | 1 |
| IDH2 | 1 | 1 |
| Equivocal | 1 | 0 |
| Case No. | PNA clamping | Direct sequencing | DIA-H09 | Race | Age (yr) | Sex | Site | F/U (mo) | Met |
|---|---|---|---|---|---|---|---|---|---|
| 1 | R132 | R132H | (+) | Korea | 47 | M | Face | NED 32 | No |
| 2 | R132 | Wild | (–) | Brazil | 12 | F | Foot | DOD 48 | No |
| 3 | R172 | Wild | (–) | Korea | 14 | F | Ilium | NED 72 | No |
| 4 | Equivocal | R172K | (–) | Korea | 19 | M | Femur | NED 48 | No |
| Characteristic | Gene profile |
||
|---|---|---|---|
| Wild | Mutant | p-value | |
| Race | |||
| Argentina | 12 | 0 | .009 |
| Brazil | 37 | 1 | |
| Korea | 8 | 3 | |
| Age (yr) | |||
| < 20 | 29 | 3 | .614 |
| > 20 | 28 | 1 | |
| Sex | |||
| Female | 24 | 2 | > .999 |
| Male | 33 | 2 | |
| Tumor site | |||
| Central | 21 | 1 | > .999 |
| Peripheral | 36 | 3 | |
| Distant metastasis | |||
| Yes | 9 | 0 | .554 |
| No | 25 | 4 | |
PNA, pentose nucleic acid.
R132H, CGT > CAT; R172K, AGG > AAG. PNA, pentose nucleic acid; F/U, follow-up; Met, metastasis; M, male; NED, no evidence of disease; F, female; DOD, dead of disease.

