 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 56(6); 2022 > Article
-
Newsletter
What’s new in soft tissue and bone pathology 2022–updates from the WHO classification 5th edition -
Erica Y. Kao,1
 , Jose G. Mantilla2
, Jose G. Mantilla2 -
Journal of Pathology and Translational Medicine 2022;56(6):385-386.
DOI: https://doi.org/10.4132/jptm.2022.10.18
Published online: November 15, 2022
1Department of Pathology, Brooke Army Medical Center, San Antonio, TX, USA
2Department of Pathology, University of Washington, Seattle, WA, USA
- Corresponding Author: Erica Y. Kao, MD Department of Pathology, Brooke Army Medical Center, San Antonio, Texas, USA E-mail: erica.kao.mil@health.mil
- This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com.
• Received: September 20, 2022 • Accepted: October 18, 2022
© 2022 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- The 2020 release of the WHO Classification of Soft Tissue and Bone Tumors, 5th edition, contains several changes driven by new knowledge in the field. These include reclassification of some entities, refinement of risk classification systems, and the inclusion of novel disease processes, many of which are driven by recurrent gene fusions. The most notable changes are described here.
- Lipomatous tumors
- • Atypical spindle cell/pleomoprhic lipomatous tumor
- - Composed of variable proportions of atypical spindle cells, adipocytes, univacuolated or multivacuolated lipoblasts, pleomorphic to multinucleated cells, and myxoid to collagenous stroma.
- - Lack of MDM2 or CDK4 amplification.
- - Rb expression is generally lost.
- - Low rate of local recurrence (10%–15%); no known risk of dedifferentiation.
- • Myxoid pleomorphic liposarcoma
- - Occurs predominantly in children and young adults with a predilection for the mediastinum.
- - Admixture of areas resembling myxoid liposarcoma with more cellular areas containing overt nuclear pleomorphism, which resembles pleomorphic liposarcoma.
- - Lacks recurrent chromosomal changes, namely MDM2 amplification and DDIT3 gene fusion.
- - Clinical behavior akin to pleomorphic liposarcoma.
- Fibroblastic/myofibroblastic tumors
- • EWSR1::SMAD4 positive fibroblastic tumor
- - Small dermal and subcutaneous acral nodule, with indolent biological behavior.
- - Histologic zonation with acellular hyalinized center and peripheral fascicular monomorphic spindle cell growth.
- - Diffuse ERG nuclear expression in the absence of CD34 and SMA expression.
- - EWSR1::SMAD4 fusion.
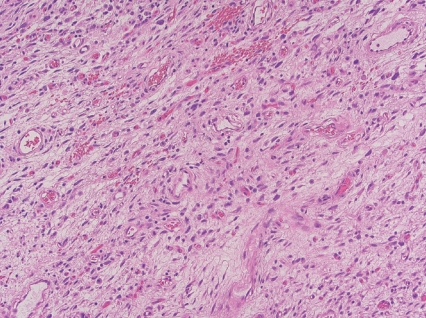
- • Angiofibroma of soft tissue
- - Benign neoplasm with rare local recurrence.
- - Uniformly bland short spindle cells in variably myxoid to collagenous stroma, with prominent vascular network of small thin-walled branching blood vessels (Fig. 1).
- - NCOA2 gene rearrangements in up to 80%.
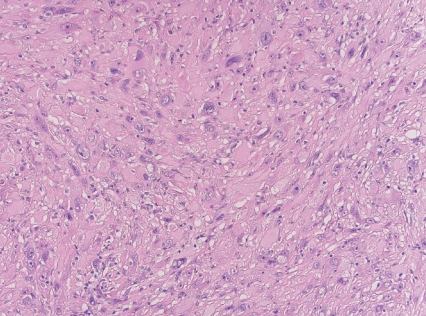
- • Superficial CD34-positive fibroblastic tumor
- - Rare, slow-growing, indolent neoplasm.
- - Superficial location, typically in the lower extremities.
- - Large eosinophilic cells with granular to glassy cytoplasm; marked pleomorphism with low mitotic count (Fig. 2).
- - CD34 and frequent keratin expression.
- Smooth muscle tumors
- • Inflammatory leiomyosarcoma
- - Rare, and thought to be relatively indolent compared to conventional leiomyosarcoma.
- - Typically arise in the deep extremities.
- - Variably atypical eosinophilic spindle cells in fascicles, with mitotic activity and prominent, usually diffuse, mixed (predominantly mononuclear) inflammatory infiltrate.
- - Near-haploid karyotype.
- • EBV-associated smooth muscle tumor
- - Associated with EBV infection, usually in the setting of immunosuppression.
- - Can arise in any anatomic location, most often in visceral sites and CNS.
- - Prognosis depends on the patient’s immune condition. Most tumors do not metastasize.
- - Cytologic atypia is highly variable. In half of cases, a second population of more primitive appearing round cells are seen. T-cell inflammatory infiltrates are common.
- - Invariably positive expression of EBER.
- Vascular tumors
- • Anastomosing hemangioma
- - Benign vascular neoplasm. Often arises in viscera and can be multifocal.
- - Thin-walled anastomosing vessels lined by hobnail endothelial cells. Vascular thrombi are typical. Loosely lobulated architecture with focal infiltration into adjoining tissue. May be associated with a medium-caliber vessel.
- - Activating mutations in GNAQ or GNA14.
- • Epithelioid hemangioendotehlioma with YAP1::TFE3 gene fusion
- - Considered to have a generally more aggressive behavior.
- - Tends to have more solid growth and be vasoformative, compared to cases harboring WWTR::CAMTA1 fusion.
- Tumors of uncertain differentiation
- • Kinase gene-rearranged spindle cell neoplasms
- - Outside of infantile fibrosarcoma, this represents an emerging group of tumors with a wide morphologic spectrum.
- - Most tumors have co-expression of S100 and CD34 in the absence of SOX10.
- - They can resemble lipofibromatosis or can be composed of monomorphic spindle cells with prominent collagen deposition and hyalinization; amianthoid-like fibers and infiltrative growth may also be present.
- - Harbor gene fusions involving kinase genes, such as NTRK, ALK, RAF1 and BRAF.
- Undifferentiated round cell sarcomas
- Due to the recent expansion in molecular studies, multiple recurrent gene fusions have been described in previously unclassified round cell sarcomas. These lesions have been shown to have particular clinical and morphologic features. These include:
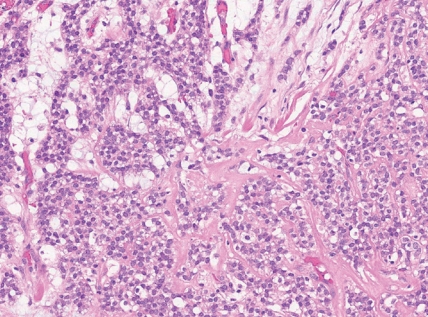
- • Round cell sarcoma with EWSR1::non-ETS fusions
- - Round and spindle cell sarcomas with fusions in EWSR1 or FUS with partners unrelated to the ETS gene family, most commonly NFATC2 or PATZ1 genes.
- - Tumors with EWSR1::NFATC2 consist of round or spindled cells arranged in cords, nests and trabeculae in a myxohyaline background (Fig. 3). They may have focal dot-like keratin and CD138 expression. They may morphologically mimic myoepithelial neoplasms or ossifying fibromyxoid tumor, among others.
- - Sarcomas with EWSR1::PATZ1 fusion have a broad morphologic spectrum. They may have round to spindle cells within a fibrous stroma. Co-expression of myogenic and nerve sheath markers has been described.
- • CIC-rearranged sarcoma
- - Round cell sarcoma characterized by CIC gene fusions, most commonly CIC::DUX4. Other fusion partners include FOXO4, LEUTX, NUTM1 and NUTM2A.
- - Composed of sheets of large round cells with mild nuclear pleomorphism, lightly eosinophilic cytoplasm, geographic necrosis and brisk mitotic activity (Fig. 4).
- - Immunohistochemically, they show variable CD99 expression, nuclear WT1 and DUX4 reactivity.
- - Response to chemotherapy is poor compared to Ewing sarcoma and BCOR fusion sarcomas.
- • Sarcoma with BCOR alterations
- - Primitive round cell sarcoma with BCOR gene fusions, most commonly BCOR::CCNB3, followed by BCOR internal tandem duplication.
- - Composed of primitive round to spindled cells in nests, sheets or fascicles in variably myxoid stroma, which may morphologically resemble synovial sarcoma. Their clinical response to chemotherapy is favorable when compared to Ewing sarcoma and CIC-fusion sarcomas.
- - Sarcomas with BCOR fusion are slightly more common in bone and tend to arise in patients younger than 20 years. On the other hand, sarcomas with BCOR internal tandem duplication tend to arise in the soft tissues of the trunk, retroperitoneum, and head and neck. These usually occur within the first year of life or may present at birth.
SELECT NEW ENTITIES
- • Mammary-type myofibroblastoma has been renamed as myofibroblastoma.
- • Melanotic schwannoma has been renamed to malignant melanotic nerve sheath tumor. This change better reflects its aggressive clinical behavior.
CHANGES IN NOMENCLATURE
- • Solitary fibrous tumor
- - Risk stratification based on age, tumor size, mitotic activity, and necrosis is recommended [1].
- • Spindle cell/sclerosing rhabdomyosarcoma
- - Subclassification depends on the presence of genetic alterations associated with prognosis.
- - Tumors with VGL2L, NCOA2 and CITED2 gene rearrangements typically arise in infants and have favorable prognosis.
- - Tumors with MYOD1 mutations usually arise in adolescents and adults, and have unfavorable prognosis.
- - Tumors with TFCP2/NCOA2 gene rearrangements can be intraosseous.
OTHER CHANGES
- Dr. Kao has been an author for PathologyOutlines since 2020. She is currently a staff pathologist at Brooke Army Medical Center in San Antonio, TX in where she practices Bone and Soft Tissue pathology and is an Assistant Professor for the Uniformed Services University of the Health Sciences. She obtained her M.D. at the Uniformed Services University of the Health Sciences in Bethesda, MD, completed her residency in AP/CP Pathology at Brooke Army Medical Center, and fellowship in Bone and Soft Tissue Pathology at the University of Washington.
- Dr. Mantilla has been an author for PathologyOutlines since 2019 and part of the PathologyOutlines editorial board since December 2020. He is an Assistant Professor of Laboratory Medicine and Pathology and Program Director of the Surgical Pathology fellowship at the University of Washington in Seattle. He obtained his M.D. at the Pontificia Universidad Javeriana (Bogotá, Colombia), followed by a residency in Anatomic and Clinical Pathology at Montefiore Medical Center in New York and fellowships in Surgical Pathology and Bone and Soft Tissue Pathology at the University of Washington.
Meet the Authors
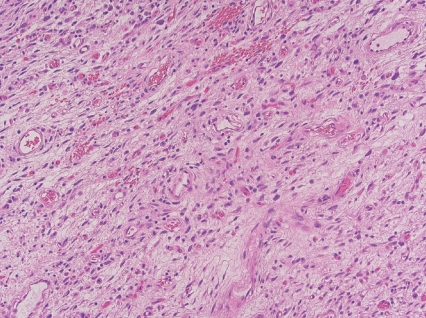
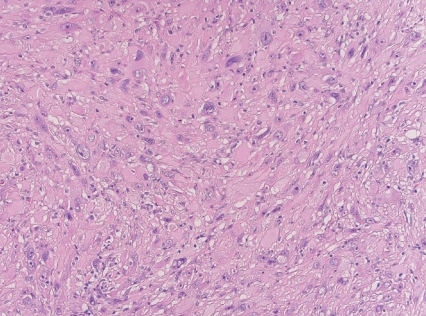
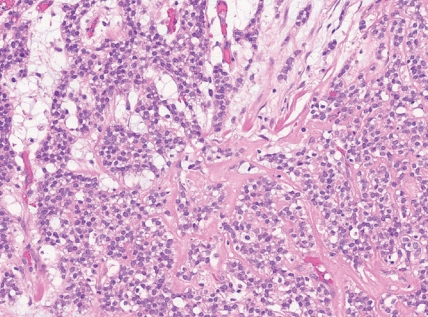
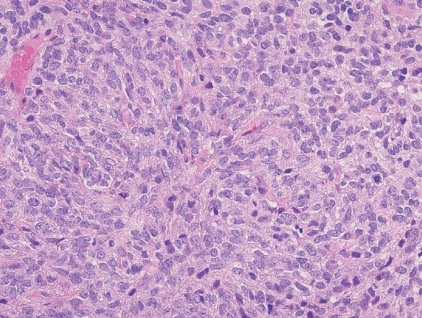
Figure & Data
References
Citations
Citations to this article as recorded by 
- Chondroblastic Osteosarcoma in an Adolescent: Are Conventional Biopsy Techniques Just Scratching the Surface?
Neeraj Kumar Dhiman, Arjun Mahajan, Trupti Jain, Ajit Kumar Vishwakarma, Rahul Agrawal
Indian Journal of Otolaryngology and Head & Neck Surgery.2025; 77(2): 1023. CrossRef - Radiomics and Deep Learning Model for Benign and Malignant Soft Tissue Tumors Differentiation of Extremities and Trunk
Miaomiao Yang, Xiuming Zhang, Jiyang Jin
Academic Radiology.2025; 32(5): 2838. CrossRef - Conference on challenges in sarcoma (CCS) 2024: Expert opinions on non-evidence-based management aspects
Silvia Hofer, Chantal Pauli, Beata Bode, Sylvie Bonvalot, Christina Fotopoulou, Hans Gelderblom, Rick L Haas, Jendrik Hardes, Peter Hohenberger, Jens Jakob, Wolfgang G. Kunz, Andreas Leithner, Bernadette Liegl-Atzwanger, Lars Lindner, Aisha Miah, Peter Re
European Journal of Cancer.2025; 220: 115368. CrossRef - European standard clinical practice recommendations for children and adolescents with Rhabdomyosarcoma a joint EpSSG, CWS and ERN PaedCan project
Johannes H.M. Merks, Eva Brack, Martin Ebinger, Véronique Minard-Colin, Anne-Sophie Defachelles, Raquel Hladun, Timothy Rogers, Jörg Fuchs, Jan Godzinski, Sheila Terwisscha van Scheltinga, Gabriela Guillén Burrieza, Henry Mandeville, Beate Timmermann, Raq
EJC Paediatric Oncology.2025; 5: 100229. CrossRef - Management of a rapidly enlarging supraclavicular mass of unknown aetiology
Mateo Cukman, Karla Luzaic, Kristina Krstanovic, Sinisa Stevanovic
BMJ Case Reports.2024; 17(2): e255774. CrossRef - Pathogenetic and molecular classifications of soft tissue and bone tumors: A 2024 update
Andrei Ionut Patrichi, Simona Gurzu
Pathology - Research and Practice.2024; 260: 155406. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-

- Related articles
-
- What’s new in neuropathology 2024: CNS WHO 5th edition updates
- What’s new in adrenal gland pathology: WHO 5th edition for adrenal cortex
- What’s new in thyroid pathology 2024: updates from the new WHO classification and Bethesda system
- What’s new in dermatopathology 2023: WHO 5th edition updates
- What’s new in hematopathology 2023: updates on mature T-cell neoplasms in the 5th edition of the WHO classification
What’s new in soft tissue and bone pathology 2022–updates from the WHO classification 5th edition




Fig. 1. Angiofibroma of soft tissue.
Fig. 2. Superficial CD34-positive fibroblastic tumor.
Fig. 3. Round cell sarcoma with EWSR1::non-ETS fusions.
Fig. 4. CIC-rearranged sarcoma.
Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
What’s new in soft tissue and bone pathology 2022–updates from the WHO classification 5th edition

