 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 56(6); 2022 > Article
-
Newsletter
What’s new in kidney tumor pathology 2022: WHO 5th edition updates -
Maria Tretiakova,

-
Journal of Pathology and Translational Medicine 2022;56(6):383-384.
DOI: https://doi.org/10.4132/jptm.2022.08.16
Published online: September 8, 2022
Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA, USA
- Corresponding Author: Maria Tretiakova, MD, PhD Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA, USA E-mail: mariast@uw.edu
- This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com
• Received: August 13, 2022 • Accepted: August 16, 2022
© 2022 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- The 5th edition WHO Classification of Urinary and Male Genital Tumours (2022) introduces significant changes relevant to daily practice, especially in the completely restructured renal cell tumor chapters. Herein we highlight the most important diagnostic updates of known kidney tumor types, new and molecularly defined entities and emerging/provisional entities.
- Papillary renal cell carcinoma (PRCC)
- • Subclassification into type 1 and type 2 is no longer recommended.
- • PRCC has classic morphology (papillae with vascular cores, foamy histiocytes and psammoma bodies) but can exhibit other appearances, including predominant solid phenotype, biphasic pattern with squamoid alveolar cells, eosinophilic cells with brisk inflammation mimicking Warthin tumor or predominant vacuolated cells mimicking clear cell RCC.
- • Many tumors previously diagnosed as type 2 PRCC now constitute independent entities.
- Clear cell papillary renal cell tumor (CCPRCT)
- • Renamed from carcinoma to tumor due to uniformly indolent behavior.
- • Low-stage, low-grade tumor with tubulopapillary and cystic architecture composed of clear cells with linearly aligned luminally oriented nuclei.
- • Co-express CK7 and CAIX (cup-like), often positive for HMWCK, but negative for CD10, and lack recurrent cytogenetic abnormalities or VHL gene alterations.
- Chromophobe RCC (ChRCC)
- • ChRCC can have non-conventional morphology with trabecular, alveolar, papillary, microcystic or cystic architecture, but all these phenotypes typically maintain CK7/CKIT co-expression, characteristic chromosomal monosomies and favorable prognosis.
- Diagnostic recommendations
- • An unequivocal diagnosis of multilocular cystic neoplasm of low malignant potential (MCNLMP), CCPRCT and oncocytoma should not be made on needle biopsy alone because of limited sampling and overlapping features with malignant counterparts.
UPDATES IN ESTABLISHED RENAL TUMORS
- This heterogeneous group of tumors often shows significant morphologic overlap with other renal tumors. Definitive diagnosis requires molecular studies like NGS, RNAseq, FISH or RT-PCR [1].
- TFE3-rearranged RCC (formerly named MiTF family Xp11 translocation RCC)
- • Heterogeneous tumors in younger patients with mixed papillary and solid architecture, psammoma bodies and clear to eosinophilic cytoplasm.
- • Express nuclear TFE3 and variably melanocytic markers and cathepsin K.
- • TFE3 rearrangement with > 20 different gene partners creates fusion subtypes with variable tumor morphology, immunoprofile and clinical behavior.
- TFEB-rearranged RCC
- • Tumor has either translocation or amplification of TFEB on t(6;11).
- • TFEB-translocation RCC is a low-stage indolent biphasic neoplasm with nests of large clear cells and smaller cells clustered around basement membrane material.
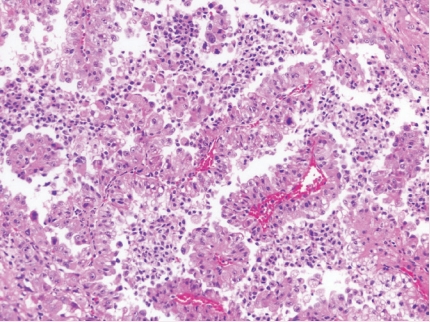
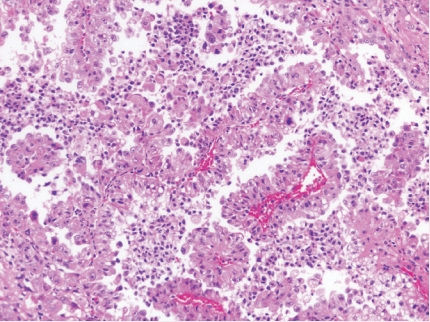
- • TFEB-amplified RCC is an often high-grade and high-stage tumor with frequent oncocytic and papillary morphology affecting older patients (Fig. 1).
- • Both subtypes consistently express nuclear TFEB, cathepsin K and melanocytic markers.
- ELOC (formerly TCEB1)-mutated RCC (novel entity)
- • Uncommon indolent clear cell tumor with solid and papillary growth patterns and nodular appearance due to traversing fibromuscular bands and septa.
- • Morphologically mimic conventional clear cell and tuberous sclerosis-associated RCCs.
- • Consistently immunoreactive for CK7 and can be focally positive for HMWCK.
- • Develops due to bi-allelic inactivation of TCEB1 (ELOC) on chromosome 8 encoding for elongin C of the VHL complex with intact VHL and mTOR pathway genes.
- Fumarate hydratase (FH)-deficient RCC
- • Renamed from hereditary leiomyomatosis-associated RCC.
- • Aggressive tumor with mixed papillary, solid, tubulocystic and cribriform architecture, composed of high-grade cells with cherry-red macronucleoli.
- • Germline (majority of cases) or somatic FH gene mutations should be suspected with immunostaining demonstrating FH protein loss and/or 2-succinocysteine (2SC) gain.
- Succinate dehydrogenase (SDH)-deficient RCC
- • Rare tumor with distinct solid morphology of bland eosinophilic cells with bubbly inclusions.
- • Loss of SDHB protein expression and germline mutation in SDH gene complex.
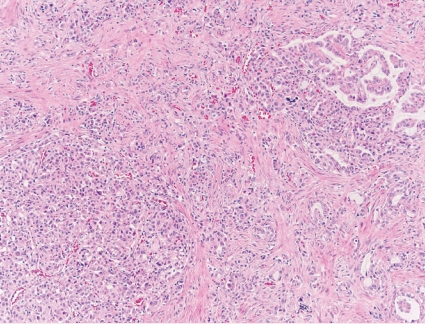
- ALK-rearranged RCC (novel entity)
- • Very rare group of extremely heterogeneous eosinophilic tumors which develop due to fusions of anaplastic lymphoma kinase gene (ALK) at 2p23 resulting in ALK protein overexpression.
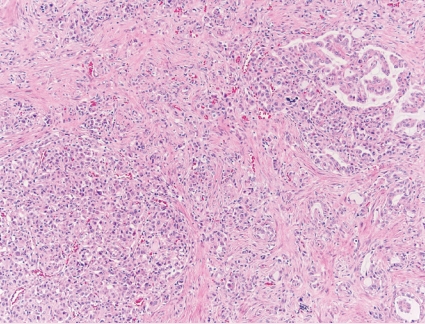
- • May show cytoplasmic vacuolization; solid, papillary or cribriform architecture with mucin production; psammoma bodies; metanephric-like, rhabdoid or spindle cell morphology (Fig. 2).
- SMARCB1-deficient renal medullary carcinoma
- • Renamed from renal medullary carcinoma.
- • Highly aggressive medulla-centered adenocarcinoma predominantly affecting patients with sickle cell trait (hemoglobinopathy) and of African ancestry.
- • Presents as locally advanced or metastatic disease with fast-growing infiltrating tumor composed of cords, nests, tubules and cribriform structures in desmoplastic background with brisk mitoses.
- • Loss of SMARCB1 (INI1, SNF5, BAF47) protein expression on immunostaining reflects inactivation of SMARCB1 at 22q11.23 by chromosome translocations or deletions.
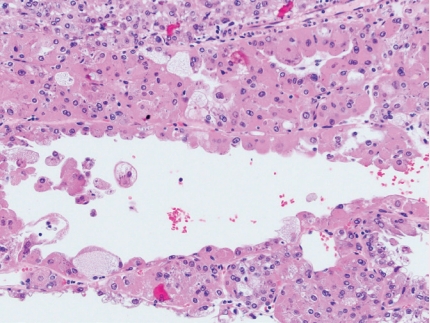
- Eosinophilic solid and cystic RCC
- • Novel distinct entity in “OTHER RENAL TUMORS” category
- • Originally described in patients with tuberous sclerosis complex, but can occur sporadically due to TSC1 or TSC2 mutations.
- • Indolent tumor disproportionately affecting women, with only rare reported metastases.
- • Solid and cystic architecture, voluminous eosinophilic cytoplasm and coarse basophilic granularity.
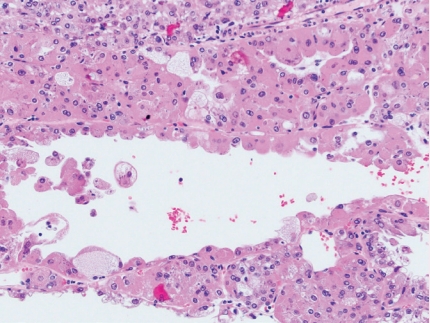
- • CK20 and cathepsin K are positive and there is a lack of CK7/CKIT expression (Fig. 3).
NEW CATEGORY OF MOLECULARLY DEFINED RENAL TUMORS
- • These tumor types are still not part of classification, but discussed in WHO [2].
- Thyroid-like follicular carcinoma
- • Rare kidney tumor composed of tightly packed follicle-like cysts filled with eosinophilic colloid-like material and lined by cuboidal cells with scant cytoplasm and oval to round nuclei.
- • Positive for PAX8, CK19 and CK7; negative for TTF1 and thyroglobulin.
- • Recurrent EWSR1::PATZ1 fusion.
- Other oncocytic tumors (oncocytic tumor, NOS)
- • These are heterogeneous groups of tumors that do not fulfill criteria for oncocytoma or eosinophilic variant of chromophobe RCC (or other specific entities).
- • Hybrid oncocytic chromophobe tumor (HOCT) is an indolent oncocytic neoplasm with borderline (intermediate) features between oncocytoma and ChRCC. It can be solitary and sporadic, but in Birt-Hogg-Dubé syndrome often is multifocal and bilateral, exhibiting a checkerboard mosaic pattern and harboring mutations in FLCN.
- • Eosinophilic vacuolated tumor (EVT) is characterized by solid growth, cytoplasmic vacuolization, entrapped tubules and large vessels, prominent nucleoli, CK7-/CKIT+ immunoprofile and mutations in mTOR pathway genes.
- • Low-grade oncocytic tumor (LOT) is a solid neoplasm with bland low-grade nuclei, CK7+/CKIT- immunoprofile and mutations in mTOR pathway genes.
- Biphasic hyalinizing psammomatous RCC
- • Rare biphasic tumor with larger cells forming tubules, papillae and acini and smaller cells clustered around hyalinized basement membrane material in glomeruloid or nested pattern.
- • It has sclerotic stroma with abundant psammoma bodies and bi-allelic loss of NF2.
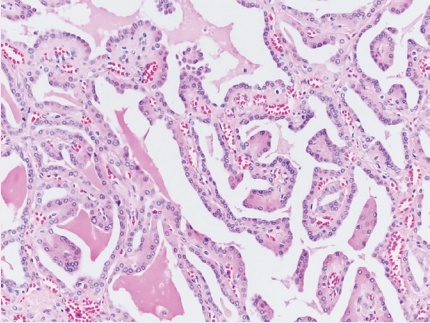
- Papillary renal neoplasm with reverse polarity
- • Formerly considered as a subtype of PRCC.
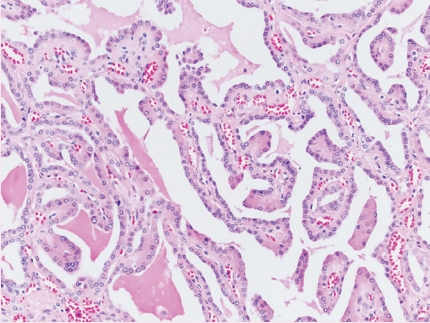
- • Eosinophilic tumor with branching papillary architecture and reverse polarity of low-grade nuclei (Fig. 4).
- • Positive for GATA3, negative for vimentin and variable for AMACR.
- • Has recurrent mutations of KRAS and lacks trisomy 7/17.
EMERGING/PROVISIONAL ENTITIES
- Dr. Tretiakova has been an author for PathologyOutlines since 2015, part of the PathologyOutlines editorial board since 2019, and Deputy Editor in Chief for GU Pathology since 2021. She is currently a Professor at the University of Washington where she primarily practices GU Pathology. Dr. Tretiakova is an international expert in GU pathology and has co-authored several topics in the WHO 2022 Blue Book.
Meet the Authors
- 1. Trpkov K, Hes O, Williamson SR, et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol 2021; 34: 1392-424. ArticlePubMedPDF
- 2. Trpkov K, Williamson SR, Gill AJ, et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol 2021; 34: 1167-84. ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Convolutional neural networks for the differentiation between benign and malignant renal tumors with a multicenter international computed tomography dataset
Michail E. Klontzas, Georgios Kalarakis, Emmanouil Koltsakis, Thomas Papathomas, Apostolos H. Karantanas, Antonios Tzortzakakis
Insights into Imaging.2024;[Epub] CrossRef - PI3K/AKT/mTOR Dysregulation and Reprogramming Metabolic Pathways in Renal Cancer: Crosstalk with the VHL/HIF Axis
Silviu Constantin Badoiu, Maria Greabu, Daniela Miricescu, Iulia-Ioana Stanescu-Spinu, Radu Ilinca, Daniela Gabriela Balan, Andra-Elena Balcangiu-Stroescu, Doina-Andrada Mihai, Ileana Adela Vacaroiu, Constantin Stefani, Viorel Jinga
International Journal of Molecular Sciences.2023; 24(9): 8391. CrossRef - Machine Learning Integrating 99mTc Sestamibi SPECT/CT and Radiomics Data Achieves Optimal Characterization of Renal Oncocytic Tumors
Michail E. Klontzas, Emmanouil Koltsakis, Georgios Kalarakis, Kiril Trpkov, Thomas Papathomas, Apostolos H. Karantanas, Antonios Tzortzakakis
Cancers.2023; 15(14): 3553. CrossRef - Serum Oxidative and Nitrosative Stress Markers in Clear Cell Renal Cell Carcinoma
Sabina Galiniak, Marek Biesiadecki, Mateusz Mołoń, Patrycja Olech, Krzysztof Balawender
Cancers.2023; 15(15): 3995. CrossRef - Redefining Renal Cell Carcinoma: A Molecular Perspective on Classification and Clinical Implications
Arjun Athreya Raghavan, Ian W Gibson, Robert Wightman, Piotr Czaykowski, Jeffrey Graham
European Medical Journal.2023; : 116. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
- Related articles
-
- What’s new in neuropathology 2024: CNS WHO 5th edition updates
- What’s new in adrenal gland pathology: WHO 5th edition for adrenal cortex
- What’s new in genitourinary pathology 2023: WHO 5th edition updates for urinary tract, prostate, testis, and penis
- What’s new in dermatopathology 2023: WHO 5th edition updates
- What’s new in hematopathology 2023: updates on mature T-cell neoplasms in the 5th edition of the WHO classification
What’s new in kidney tumor pathology 2022: WHO 5th edition updates




Fig. 1. TFEB-amplified RCC.
Fig. 2. ALK-rearranged RCC.
Fig. 3. Eosinophilic solid and cystic RCC.
Fig. 4. Papillary renal neoplasm with reverse polarity.
Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
What’s new in kidney tumor pathology 2022: WHO 5th edition updates

