 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 46(5); 2012 > Article
-
Original Article
A Proposal for Creating a Guideline for Cancer Registration of the Fibromatosis, PEComa Group, Malignant LymphomaIn Situ and Dendritic Cell Tumors (III) - Changyoung Yoo1,2,, Chang Suk Kang3,4,, Yoon La Choi2,5, Hye Yoon Kang2,6, Jin Man Kim4,7,8, Young Hye Koh4,5, Joo Hee Lee4,9, Seung Sook Lee4,10, In Sun Kim4,11, Dong Hoon Kim2,12, Yong Ku Park2,9, Jin Hee Sohn8,12
-
Korean Journal of Pathology 2012;46(5):436-442.
DOI: https://doi.org/10.4132/KoreanJPathol.2012.46.5.436
Published online: October 25, 2012
1Department of Pathology, St. Vincent's Hospital, The Catholic University of Korea College of Medicine, Suwon, Korea.
2The Society of Bone and Soft Tissue Study Group, Seoul, Korea.
3Department of Pathology, Yeouido St. Mary's Hospital, The Catholic University of Korea College of Medicine, Seoul, Korea.
4The Korean Study Group of Hematopathology, Seoul, Korea.
5Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
6Department of Pathology, Bundang CHA Hospital, CHA University College of Medicine, Seongnam, Korea.
7Department of Pathology, Chungnam National University School of Medicine, Daejeon, Korea.
8The Cancer Registration Committee of the Korean Society of Pathologist, Seoul, Korea.
9Department of Pathology, Kyung Hee University School of Medicine, Seoul, Korea.
10Department of Pathology, Korea Cancer Center Hospital, Seoul, Korea.
11Department of Pathology, Korea University School of Medicine, Seoul, Korea.
12Department of Pathology, Kangbuk Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- Corresponding Author: Jin Hee Sohn, M.D. Department of Pathology, Kangbuk Samsung Medical Center, Sungkyunkwan University School of Medicine, 29 Saemunan-ro, Jongno-gu, Seoul 110-746, Korea. Tel: +82-2-2001-2391, Fax: +82-2-2001-2398, jhpath.sohn@samsung.com
*Changyoung Yoo and Chang Suk Kang contributed equally to this work.
© 2012 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 10,781 Views
- 55 Download
- 1 Scopus
Abstract
-
Background
- Understanding the biologic behavior of a tumor is a prerequisite for tumor registration code assignment. The aim of this report was to propose appropriate behavior codes of the International Classification of Disease Oncology 3 (ICD-O3) to rare, yet pathologically interesting hematopoietic and soft tissue tumors.
-
Methods
- The Study Group for Hematopathology, the Bone and Soft Tissue Pathology Study Group, and the Cancer Registration Committee prepared the questionnaire containing provisional behavior codes of selected diseases.
-
Results
- In situ lesions of mantle cell and follicular lymphomas, dendritic cell tumors, and neoplasms with perivascular epithelioid cell differentiation (PEComa), not otherwise specified were classified as malignant (-/3). The fibromatosis group, with the exception of lipofibromatosis, was proposed as benign (-/0). Lipofibromatosis and several diseases that belong to the PEComa group were proposed as uncertain malignant potential (-/1). For the hematologic and soft tissue tumors, 274 and 288 members of the Korean Society of Pathologists, respectively, provided opinions through questionnaire, and most responders showed agreement with the provisional behavior code proposed.
-
Conclusions
- The determination of behavior codes for the rare diseases described in this study, especially those of the PEComa group or malignant lymphoma, could be viewed as impractical and premature, but this study provides the basis for future research on this topic.
- Selection of tumors
- After several conferences and workshops, each study group selected the tumors and determined the provisional behavior codes. The Study Group for Hematopathology selected in situ lesions of malignant lymphomas, and histiocytic lesions for discussion. In situ lesions included mantle cell lymphoma in situ and follicular lymphoma in situ, and histiocytic lesions included follicular dendritic cell tumors, interdigitating dendritic cell tumors, and Langerhans cell histiocytosis.
- The Bone and Soft Tissue Pathology Study Group initially selected diseases that do not have an ICD-O3 code among the bone and soft tissue tumors included in the WHO classification, including:6 fibromatosis colli, juvenile hyaline fibromatosis, inclusion body fibromatosis, superficial fibromatosis, lipofibromatosis, hyalinizing spindle cell tumor, neoplasm with perivascular epithelioid cell differentiation (PEComa), and clear cell myomelanocytic tumor among the soft tissue tumors, and aneurysmal bone cyst, fibrous dysplasia, osteofibrous dysplasia, and Erdheim-Chester disease among bone tumors. PEComa were further divided into PEComa, not otherwise specified (PEComa NOS), angiomyolipoma (AML), lymphangioleiomyomatosis (LAM), clear cell "sugar" tumor of the lung (CCST), and clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres (CCMMT). Their biologic behaviors were discussed separately. Among the selected tumors, superficial fibromatosis was excluded from this study due to its obvious benign biologic behavior. Hyalinizing spindle cell tumor was also excluded because it is a variant of low grade fibromyxoid sarcoma, and thus should be regarded as a malignant disease. Under the heading of AML, we excluded renal AML, because its biologic behavior is well known to be benign even though sarcomatous change is rarely reported.7 Aneurysmal bone cyst, fibrous dysplasia/osteofibrous dysplasia, and Erdheim-Chester disease were initially included in the discussion, but finally excluded due to well-known benignancy or rarity.
- A proposal for provisional behavior codes
- The Study Group for Hematopathology determined the codes presented both in the preexisting ICD-O3 and the WHO classification 2008 for the selected tumors so that other pathologists could compare the results, because ICD-O3 does not provide a behavior code for follicular lymphoma in situ and mantle cell lymphoma in situ, but does provide behavior code -/1 for the histiocytic and dendritic cell neoplasms. This is in contrast to the WHO classification 2008, which considers them as malignant tumors.5
- In the field of bone and soft tissue tumors, three pathologists classified these tumors on the basis of previous reports and made provisional behavior codes. They prepared materials for the charged parts, and the presentation, the discussion, and the convergence of opinions were conducted during the workshop held within the Bone and Soft Tissue Study Group. Finally, provisional behavior codes were proposed for the selected tumors. Because reports regarding these diseases were insufficient in Korea, we referred to several foreign literature databases and WHO classifications to determine provisional behavior codes.
- Preparation of questionnaire and analysis of data
- The Cancer Registration Committee held additional discussions regarding provisional behavior codes provided by the Study Group for Hematopathology and the Bone and Soft Tissue Pathology Study Group. The final lists of tumors were determined and the provisional biologic behavior codes for the corresponding diseases were established. The questionnaire was made considering these decisions and distributed to the members of KSP during the Academic Congress of Pathology and Cytopathology. Data were collected and then analyzed.
MATERIALS AND METHODS
- Provisional behavior codes settled in the workshop
- The Study Group for Hematopathology suggested the behavior code -/3 (malignant) for mantle cell lymphoma in situ and follicular lymphoma in situ. For the histiocytic and dendritic cell neoplasms, the majority of the study group members agreed on the malignant behavior (Table 2).
- For the bone and soft tissue tumors, the tumors were categorized as fibromatosis or PEComa in order to avoid the complexities of the classification. Among the fibromatosis category, fibromatosis colli, juvenile hyaline fibromatosis, and inclusion body fibromatosis were described as benign, and given the behavior code -/0. Lipofibromatosis was regarded as a borderline tumor and given behavior code -/1. Among the PEComa category, CCMMT was suggested to be a benign disease and the behavior code -/0 was applied. AML, LAM, and CCST were given behavior code -/1, and PEComa NOS was given behavior code -/3 (Table 3).
- Results of the questionnaire
- A total of 274 members of the KSP provided their opinions by completing the questionnaire. Of the responders, the following percentages indicated a behavior code of -/3 (malignant) for the respective tumor types: 75.5% (207/274) for follicular lymphoma in situ, 77% (211/274) for mantle cell lymphoma in situ, 88.0% (241/274) for follicular dendritic cell tumor, 81.0% (222/274) for interdigitating dendritic cell tumor, and 74.8% (205/274) for Langerhans cell histiocytosis. The remaining members listed behavior code -/1 (uncertain malignant potential or borderline tumor) for these tumor types, with some members providing code -/2 (carcinoma in situ) and a few others suggesting that we postpone coding (Table 4).
- Regarding responses to the questionnaire on soft tissue tumors, 288 members of the KSP provided opinions. For the fibromatosis category, most responders gave behavior code -/0 (benign tumor) for fibromatosis colli, juvenile hyaline fibromatosis, and inclusion body fibromatosis, and -/1 (uncertain malignant potential or borderline tumor) for lipofibromatosis as the provision of the Bone and Soft Tissue Study Group. For the PEComa category, most responders assigned PEComa NOS to -/3 (malignant neoplasm), AML, LAM, and CCST to -/1, and CCMMT to -/0 (Table 5).
RESULTS
- The Study Group for Hematopathology focused on in situ lesions of malignant lymphomas and histiocytic lesions. Mantle cell lymphoma in situ and follicular lymphoma in situ are defined morphologically when tumor cells are restricted to the mantle zone and intrafollicular area, respectively. However when the cells of these tumors circulate in the body, they should be classified as malignant diseases.5,8-10 Because the term "in situ" can cause confusion not only to pathologists, but also to clinicians, the Study Group for Hematopathology decided to include these lesions in order to clearly indicate that "in situ lesions" are malignant.
- There were some disputes in the study group regarding histiocytic neoplasms. In the WHO classification 2008, histiocytic and dendritic cell neoplasms are classified as malignant disease with behavior code /3 of ICD-O3.5 Even before this edition, it has been suggested that Langerhans cell histiocytosis should be considered a malignant tumor because of its risk of multisystem involvement and refractoriness to treatment,11 although it is treated as a tumor of uncertain malignant potential.4 Dendritic cell neoplasms also have morphological obscurity between borderline and malignant tumors, but they were eventually defined as malignant disease without distinction, which was in contrast to the previous edition of the WHO classification. Therefore the study group decided to include these histiocytic and dendritic cell neoplasms in this study because it may be meaningful to compare behavior codes between ICD-O3 and the WHO classification 2008. The study group selected mantle cell lymphoma in situ, follicular lymphoma in situ, follicular dendritic cell tumor, interdigitating dendritic cell tumor, and Langerhans cell histiocytosis for this study.
- In the WHO classification 2001, in situ lesion of follicular lymphoma was not mentioned.4 It was only introduced in the WHO classification 2008 under the name intrafollicular neoplasia "in situ" follicular lymphoma, and defined morphologically as architecturally normal appearing lymph nodes. Other lymphoid tissues harboring one or more follicles that overexpress Bcl-2 in centrocytes and centroblasts with or without a monomorphic cytologic appearance was described to be suggestive of follicular lymphoma.5 The meaning of this tumor has not been completely established yet, but it may represent the tissue counterpart of circulating clonal B-cells possessing BCL-2 rearrangement,5,9 or the earliest evidence of true follicular lymphoma that subsequently progress to overt follicular lymphoma.5,10,12 Mantle cell lymphoma "in situ" is even mentioned in the WHO classification 2001, but the term "in situ" is only found in the WHO classification 2008.4,5 By the definition, this tumor is confined to the inner mantle zone of lymphoid tissue.5,8
- Histiocytic and dendritic cell tumors are classified as borderline tumors and are distinguished from their malignant counterpart sarcoma in the WHO classification 2001. They became evenly classified as malignant tumors without distinction of tumor or sarcoma in the WHO classification 2008.4,5 Follicular dendritic cell tumor is a rare malignant tumor originating from follicular dendritic cells. It can arise in any organ, but most commonly occurs in the cervical lymph node. They show an indolent clinical course but have frequent local recurrence and distant metastasis.5,13 Interdigitating dendritic cell tumor is also a very rare disease, and usually occurs as a solitary lesion in the lymph node, although extranodal lesions have been reported.5,14 Langerhans cell histiocytosis usually occurs as solitary lesion in children. Typical of multisystem lesions, it is refractory to treatment and associated with high mortality.5,15
- The Study Group for Hematopathology did not propose different behavior codes, but rather decided to provide both codes that are presented in the preexisting ICD-O3 and the WHO classification 2008. The biologic codes presented in the WHO classification 2001 are the same as the codes of ICD-O3. The questionnaire form produced by the Study Group for Hematopathology was presented to the members of the KSP at academic congresses. The results of the questionnaire indicated that, members of the KSP generally agreed on the biological behavior of follicular lymphoma in situ, mantle cell lymphoma in situ, follicular dendritic cell tumor, interdigitating dendritic cell tumor, and Langerhans cell histiocytosis as malignant disease, and therefore, the Study Group for Hematopathology and the Cancer Registration Committee could propose behavior code -/3 for these tumors. The study group also suggested that the diagnostic names of Langerhans cell histiocytosis, unifocal Langerhans cell histiocytosis, and multifocal Langerhans cell histiocytosis should all be recapitulated under the name of Langerhans cell histiocytosis with biologic behavior code -/3. Howerver, the final acceptance of these provisional codes may require additional studies that include long-term follow-up data, greater accumulation of knowledge, and agreement among clinicians.
- Among the bone and soft tissue tumors included in the WHO classification,6 the study group selected tumors that do not have an ICD-O3 code; however, the biologic behaviors of these tumors are well known among pathologists even though ICD-O3 codes were not granted. Of these, the following diseases were selected: fibromatosis colli, juvenile hyaline fibromatosis, inclusion body fibromatosis, superficial fibromatosis, lipofibromatosis, hyalinizing spindle cell tumor, PEComa, and clear cell myomelanocytic tumor.
- Among the tumors of bone and soft tissue presented in the WHO classification, many tumors have behavior code -/1, such as, desmoid-type fibromatosis, inflammatory myofibroblastic tumor, and myopericytoma. These tumors have some characteristics in common, such as local recurrence, somewhat poor clinical outcome, and infrequent distant metastasis.6 Therefore, these characteristics were considered in this study when determining the biologic behavior of the diseases.
- Fibromatosis colli is a benign, site-specific lesion that occurs in the distal sternocleidomastoid muscle of infants.6 It occurs in 4% of live births, and the majority of affected infants are diagnosed before 6 months of age. The site of involvement typically comprises the lower one third of the sternocleidomastoid muscle. It is managed in a non-surgical manner, but surgical intervention may be required in some patient.16,17 We considered this lesion to be a benign disease and therefore suggest a behavior code of -/0. Juvenile hyaline fibromatosis is an extremely rare and apparently non-neoplastic disorder that typically presents in infancy, and is characterized by the accumulation of extracellular "hyaline material" within skin, somatic soft tissues, and the skeleton, resulting in tumor-like masses.6 The hyaline material is produced by an aberrant population of fibroblasts. This tumor has a progressive nature and forms superficial and deep nodules with resulting deformity and dysfunction. It most frequently occurs in skin of the face and neck, gum, periarticular soft tissues, and several bone tissues. Surgery is the treatment of choice, but the recurrence rate is high.18,19 There are reports that squamous cell carcinoma can arise from juvenile hyaline fibromatosis.20,21 We considered this disease to be locally invasive with malignant potential, and therefore behavior code -/1 may be appropriate. However, definitive evidence of malignancy or metastasis was not found, and therefore we suggest biologic code -/0. Inclusion body fibromatosis is a rare benign proliferation of fibroblastic and myofibroblastic cells that typically occurs on the digits of young children. It is named for the intracytoplasmic inclusions that are detected in a minority of the lesional cells.6 Treatment is surgical excision, but the local recurrence rate is high.22,23 Thus, based on this characteristic, the biologic behavior may be /1, but because definitive evidence of malignancy or metastasis was not found, we also suggest behavior code -/0 for this disease as well. Lipofibromatosis is a rare pediatric tumor and a histologically distinctive fibrofatty tumour of childhood, which was previously designated as infantile fibromatosis of non-desmoid type with a predilection for distal extremities. The tumor has a high rate of non-destructive local recurrence, but exhibits no metastatic potential.24,25 Because of its local recurrence, we suggest biologic behavior code -/1.
- The PEComa category showed good agreement among participants and attendees of the workshop. PEComa is a neoplasm of perivascular epithelioid cells, and includes several diseases within the PEComa family.6 However, their biologic behaviors have not been established because of the rarity of the disease and uncertain criteria for malignancy. The subset of PEComa tumors with malignant behavior do show a high mitotic index, necrosis, marked cytologic atypia, and an infiltrative growth pattern.26 AML in this study represented non-renal AML. AML usually occurs in the kidney, but is also found in the liver, lung, uterus, skin and oral cavity in rare situations.27 One report has indicated that LAM has a poor prognosis due to respiratory failure.28 CCST is considered benign, but tumors larger than 2 cm that are symptomatic and focally necrotic should be regarded as potentially malignant neoplasms.29 CCMMT has predilection for children and young adults, and it is known as a relatively benign disease.30 As presented above, AML, LAM, and CCST could be considered as behavior code -/1, and CCMMT as -/0. PEComa NOS, which does not show AML, LAM, CCST, or CCMMT characteristics, is considered a subset of PEComa with malignant behavior,26 and therefore we suggest behavior code -/3 for this tumor type.
- We could not find studies that were similar to this study in literature databases from around the world, so comparison between our data and others was not possible. Malignant lymphomas are generally thought of as malignant diseases by physicians. However, new disease entities and a gradual increase of our understanding of rare diseases have resulted in a need for the continuous reconsideration of the biologic behavior of theses tumors. In Korea, the incidence of tumors of lymphoid tissue is increasing, and therefore the results of this study should be considered in the future as a comparator for newly reported studies.
- Tumors of the bone and soft tissue undergo continual changes in the concept, classification, nomination, and biologic behavior. Bone and soft tissue tumors have a relatively low incidence, and therefore experience regarding these tumors is limited, especially for the diseases selected in this study. Although the majority of diseases entities in the study were benign or had uncertain biologic behavior and were not pertinent to the purpose of cancer registration, we think believe it is of great value to discuss and provide the behavior code for these rare disease entities. Therefore, this study has provided behavior codes for these selected tumors, but as future studies provide a more detailed understanding of these tumors, a further refinement may be necessary. In addition, tumor types not included in our questionnaire will be assessed in the future.
DISCUSSION
Acknowledgments
Acknowledgments
- 1. World Health Organization (WHO), International Classification of Disease (ICD). ICD-10 Version: 2010 [Internet]. 2011; cited 2012 Mar 1. Geneva: World Helath Organization, Available from: http://apps.who.int/classifications/apps/icd/icd10online/.
- 2. Cho MY, Kang YK, Kim KM, et al. Porposal for creating a guideline for cancer registration of the gastrointestinal tumors (I). Korean J Pathol 2008; 42: 140-150.
- 3. Sohn JH, Gong G, Kim KR, et al. Proposal for creating a guideline for cancer registration of microinvasive tumors of the breast and ovary (II). Korean J Pathol 2012; 46: 226-232. ArticlePubMedPMC
- 4. Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Organization classification of tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. 2001; Lyon: IARC Press.
- 5. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 2008; 4th ed. Lyon: IARC Press.
- 6. Fletcher CD, Unni KK, Mertens F. World Health Organization classification of tumours: pathology and genetics of tumours of soft tissue and bone. 2002; Lyon: IARC Press.
- 7. Cibas ES, Goss GA, Kulke MH, Demetri GD, Fletcher CD. Malignant epithelioid angiomyolipoma ('sarcoma ex angiomyolipoma') of the kidney: a case report and review of the literature. Am J Surg Pathol 2001; 25: 121-126. PubMed
- 8. Edlefsen KL, Greisman HA, Yi HS, Mantei KM, Fromm JR. Early lymph node involvement by mantle cell lymphoma limited to the germinal center: report of a case with a novel "follicular in situ" growth pattern. Am J Clin Pathol 2011; 136: 276-281. ArticlePubMed
- 9. Jegalian AG, Eberle FC, Pack SD, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood 2011; 118: 2976-2984. ArticlePubMedPMCPDF
- 10. Henopp T, Quintanilla-Martínez L, Fend F, Adam P. Prevalence of follicular lymphoma in situ in consecutively analysed reactive lymph nodes. Histopathology 2011; 59: 139-142. ArticlePubMed
- 11. Minkov M. Multisystem Langerhans cell histiocytosis in children: current treatment and future directions. Paediatr Drugs 2011; 13: 75-86. PubMed
- 12. Montes-Moreno S, Castro Y, Rodríguez-Pinilla SM, et al. Intrafollicular neoplasia/in situ follicular lymphoma: review of a series of 13 cases. Histopathology 2010; 56: 658-662. ArticlePubMed
- 13. Pizzi M, Ludwig K, Palazzolo G, Busatto G, Rettore C, Altavilla G. Cervical follicular dendritic cell sarcoma: a case report and review of the literature. Int J Immunopathol Pharmacol 2011; 24: 539-544. ArticlePubMedPDF
- 14. Han HS, Lee OJ, Lim SN, et al. Extranodal interdigitating dendritic cell sarcoma presenting in the pleura: a case report. J Korean Med Sci 2011; 26: 304-307. ArticlePubMedPMCPDF
- 15. Matsuki E, Tsukada Y, Nakaya A, Yokoyama K, Okamoto S. Successful treatment of adult onset Langerhans cell histiocytosis with multi-drug combination therapy. Intern Med 2011; 50: 909-914. ArticlePubMed
- 16. Weiss SW, Goldblum JR. Enzinger and Weiss's soft tissue tumors. 2008; 5th ed. St. Louis: Mosby.
- 17. Canale ST, Griffin DW, Hubbard CN. Congenital muscular torticollis: a long-term follow-up. J Bone Joint Surg Am 1982; 64: 810-816. ArticlePubMed
- 18. Kan AE, Rogers M. Juvenile hyaline fibromatosis: an expanded clinicopathologic spectrum. Pediatr Dermatol 1989; 6: 68-75. ArticlePubMed
- 19. Woyke S, Domagala W, Markiewicz C. A 19-year follow-up of multiple juvenile hyaline fibromatosis. J Pediatr Surg 1984; 19: 302-304. ArticlePubMed
- 20. Kawasaki G, Yanamoto S, Mizuno A, Fujita S. Juvenile hyaline fibromatosis complicated with oral squamous cell carcinoma: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001; 91: 200-204. ArticlePubMed
- 21. Shimizu K, Ogawa F, Hamasaki Y, Murota H, Katayama I. A case of bullous pemphigoid arising in juvenile hyaline fibromatosis with oral squamous cell carcinoma. J Dermatol 2005; 32: 650-653. ArticlePubMed
- 22. Beckett JH, Jacobs AH. Recurring digital fibrous tumors of childhood: a review. Pediatrics 1977; 59: 401-406. ArticlePubMedPDF
- 23. Rimareix F, Bardot J, Andrac L, Vasse D, Galinier P, Magalon G. Infantile digital fibroma: report on eleven cases. Eur J Pediatr Surg 1997; 7: 345-348. ArticlePubMed
- 24. Fetsch JF, Miettinen M, Laskin WB, Michal M, Enzinger FM. A clinicopathologic study of 45 pediatric soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J Surg Pathol 2000; 24: 1491-1500. ArticlePubMed
- 25. Flacke S, Pauleit D, Keller E, et al. Infantile fibromatosis of the neck with intracranial involvement: MR and CT findings. AJNR Am J Neuroradiol 1999; 20: 923-925. PubMedPMC
- 26. Hornick JL, Fletcher CD. PEComa: what do we know so far? Histopathology 2006; 48: 75-82. ArticlePubMed
- 27. Knight CS, Cerfolio RJ, Winokur TS. Angiomyolipoma of the anterior mediastinum. Ann Diagn Pathol 2008; 12: 293-295. ArticlePubMed
- 28. Park HY, Nam HS, Chung MP, et al. A nationwide survey of lymphangioleiomyomatosis in Korea: recent increase in newly diagnosed patients. J Korean Med Sci 2010; 25: 1182-1186. ArticlePubMedPMCPDF
- 29. Gaffey MJ, Mills SE, Askin FB, et al. Clear cell tumor of the lung: a clinicopathologic, immunohistochemical, and ultrastructural study of eight cases. Am J Surg Pathol 1990; 14: 248-259. ArticlePubMed
- 30. Folpe AL, Goodman ZD, Ishak KG, et al. Clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres: a novel member of the perivascular epithelioid clear cell family of tumors with a predilection for children and young adults. Am J Surg Pathol 2000; 24: 1239-1246. PubMed
REFERENCES

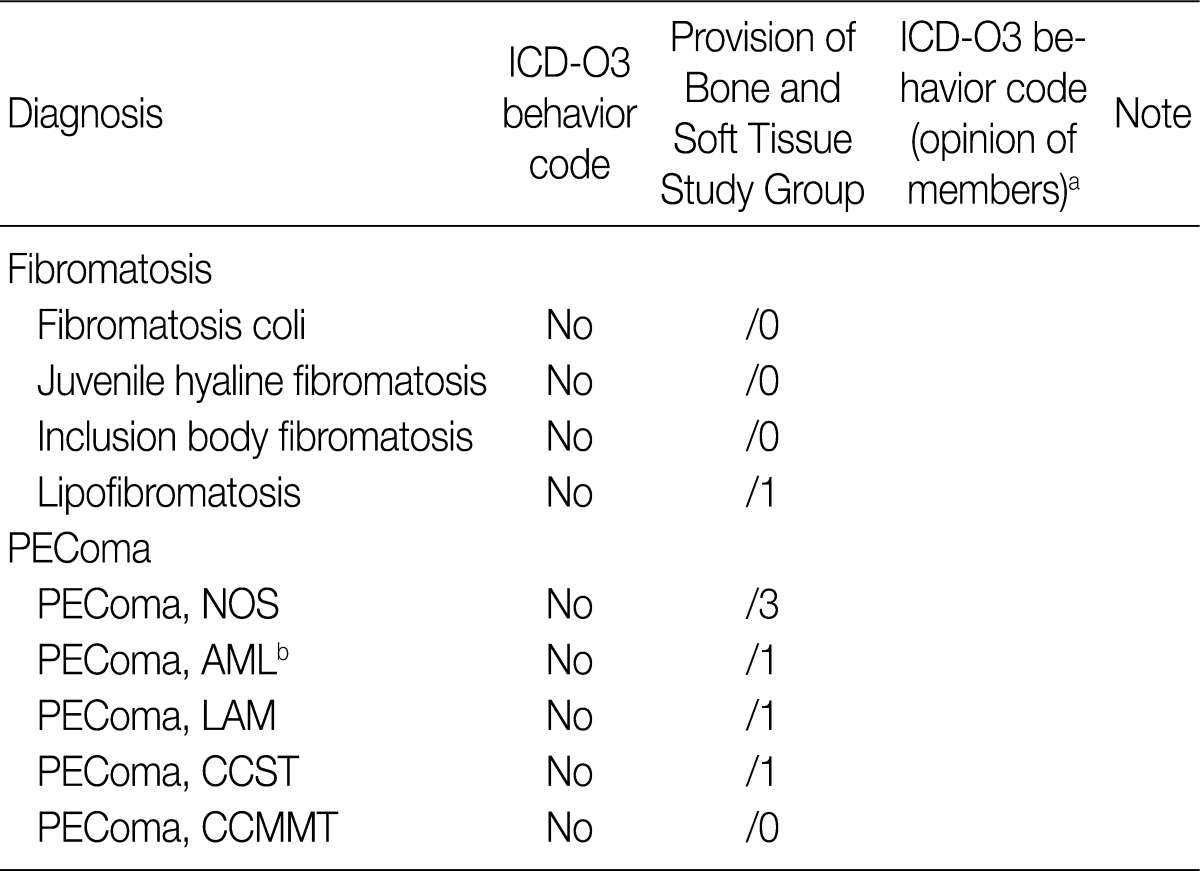
ICD-O3, International Classification of Disease Oncology 3; PEComa, perivascular epithelioid cell differentiation; NOS, not otherwise specified; AML, angiomyolipoma; LAM, lymphangioleiomyomatosis; CCST, clear cell "sugar" tumor of the lung; CCMMT, clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres.
a"Please write your opinion as /0, /1, or /3 in the blank"; bThe renal AML is excluded.

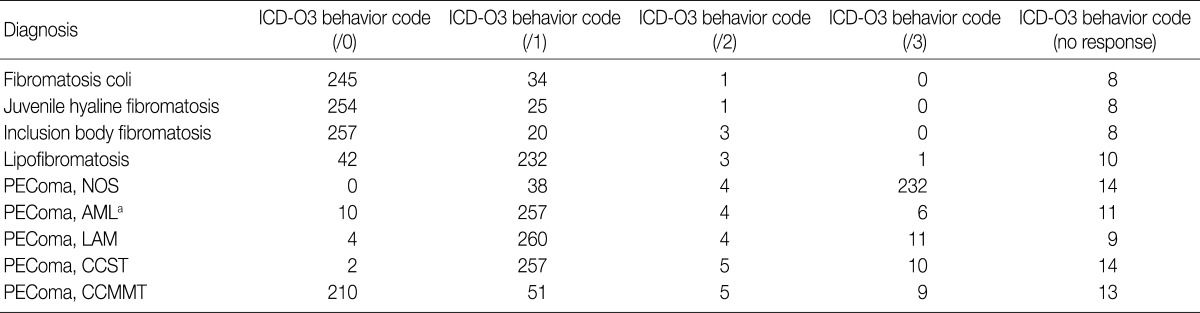
ICD-O3, International Classification of Disease Oncology 3; PEComa, perivascular epithelioid cell differentiation; NOS, not otherwise specified; AML, angiomyolipoma; LAM, lymphangioleiomyomatosis; CCST, clear cell "sugar" tumor of the lung; CCMMT, clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres.
aThe renal AML is excluded.
Figure & Data
References
Citations
 PubReader
PubReader Cite this Article
Cite this Article




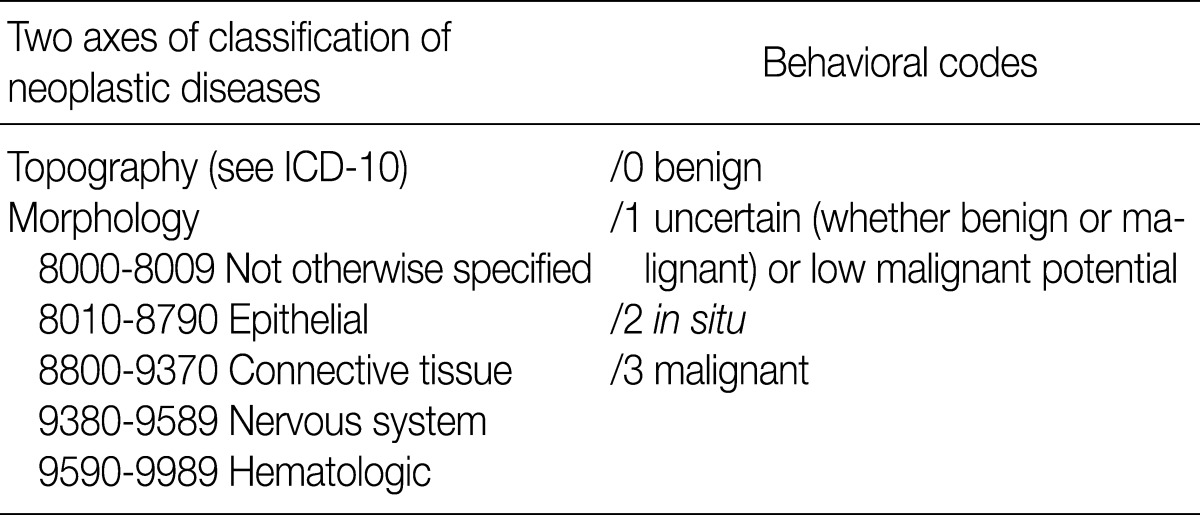
ICD-O3, International Classification of Disease Oncology 3.
ICD-O3, International Classification of Disease Oncology 3; WHO, World Health Organization.
ICD-O3, International Classification of Disease Oncology 3; PEComa, perivascular epithelioid cell differentiation; NOS, not otherwise specified; AML, angiomyolipoma; LAM, lymphangioleiomyomatosis; CCST, clear cell "sugar" tumor of the lung; CCMMT, clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres. a"Please write your opinion as /0, /1, or /3 in the blank"; bThe renal AML is excluded.
ICD-O3, International Classification of Disease Oncology 3.
ICD-O3, International Classification of Disease Oncology 3; PEComa, perivascular epithelioid cell differentiation; NOS, not otherwise specified; AML, angiomyolipoma; LAM, lymphangioleiomyomatosis; CCST, clear cell "sugar" tumor of the lung; CCMMT, clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres. aThe renal AML is excluded.

