E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 60(3); 2026 > Article
-
Review Article
Gene fusions in melanocytic lesions: an updated comprehensive review -
Volha Lenskaya1
 , Larisa Erikson2, Victor G. Prieto1, Woo Cheal Cho1
, Larisa Erikson2, Victor G. Prieto1, Woo Cheal Cho1 -
Journal of Pathology and Translational Medicine 2026;60(3):285-306.
DOI: https://doi.org/10.4132/jptm.2026.03.11
Published online: May 8, 2026
1Department of Pathology and Laboratory Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
2Department of Pathology, Children’s Hospital, University of Colorado School of Medicine, Aurora, CO, USA
- Corresponding author: Volha Lenskaya, MD, Department of Pathology and Laboratory Medicine, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 85, Houston, TX 77030, USA Tel: +1-8323505873, Fax: +1-713-745-8228, E-mail: vlenskaya@mdanderson.org
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 3,834 Views
- 127 Download
Abstract
- The scope of gene fusions in melanocytic neoplasms is broader than previously recognized, extending well beyond the Spitz-lineage neoplasms where kinase fusions involving ALK, ROS1, NTRK1/2/3, RET, MET, BRAF, and MAP3K8 define biologically and morphologically distinct tumors. Emerging studies demonstrate that a meaningful proportion of conventional non-Spitz lineage melanomas harbor oncogenic fusions. Such fusions may impact clinical behavior, histopathologic presentation and provide opportunities for targeted therapy. The World Health Organization classification of skin tumors, 5th edition, now incorporates fusion status into taxonomy and risk stratification, yet some important questions remain for further investigation: fusion-associated neoplasms can mimic non-melanocytic neoplasm; Spitz-type fusions appear in non-Spitz lesions; and melanocytic differentiation may occur in some other fusion-driven lesions. Broad-panel next-generation sequencing (including RNA-seq), together with targeted fluorescence in situ hybridization and immunohistochemistry enhances detection of known and novel fusion partners. Early clinical evidence of TRK, ALK, and ROS1 inhibitor efficacy underscores the translational promise of fusion testing and opens avenues for personalized therapy. This review synthesizes current knowledge on the genomics, histopathology, diagnosis, and therapeutic implications of fusion-driven melanocytic neoplasms, highlighting consensus points and remaining controversies.
- Recent advancements in molecular techniques, such as next-generation sequencing (NGS) and whole-transcriptome sequencing, have significantly enhanced our understanding of the tumorigenic mechanisms underlying melanocytic neoplasms, particularly through the identification of novel gene fusions. While gene fusions are commonly associated with Spitz melanocytic neoplasms, new evidence suggests that these fusion events are not limited to Spitz neoplasms. The scope of gene fusions in melanocytic tumors is broader than previously recognized. Therefore, it is increasingly important that molecular findings complement, rather than replace, the pathologist’s morphological assessment to avoid diagnostic bias and ensure accurate tumor classification. This review aims to provide a comprehensive, up-to-date overview of fusion-associated melanocytic neoplasms, emphasizing the importance of properly interpreting molecular data, which are context-dependent, as well as exploring the potential prognostic and therapeutic significance of gene fusions in these tumors (Supplementary Tables S1, S2).
INTRODUCTION
- Gene fusions are hybrid genes resulting from structural genomic rearrangements that juxtapose coding or regulatory regions of distinct genes, often resulting in aberrant transcriptional or functional activity. Most oncogenic fusions are in-frame fusions involving exonic regions of protein-coding genes that are crucial for the functioning of those proteins [1]. The most affected gene groups involved in a majority of human cancers are protein kinases (PKs) and transcription factors (TFs) [2]. The mutational dysregulation of these gene groups significantly impacts tumorigenesis due to the innate function of PKs to mediate most cellular signal transduction events by phosphorylation of specific substrates, thus modifying their activity, cellular localization, and/or association with other proteins. TFs are the “transistors” of the cellular signaling circuits, controlling the transcriptional outcome of activated signaling by binding to regulative elements of their corresponding target genes and driving or suppressing their expression [1]. Not only conventional gene fusion but precisely kinase fusions are fairly common in melanocytic lesions and seen within approximately half of all spitzoid neoplasms [3,4]. The kinase gene fusions are defined as chimeric oncogenes generated by structural rearrangements that fuse a PK domain to a heterologous partner, leading to constitutive activation of signaling pathways such as mitogen-activated protein kinase (MAPK) or phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR). The current understanding on how gene fusions drive cancer favors the aberrant gene function model rather than promoter-induced overexpression model; in other words, gene fusions act by altering protein function rather than just increasing how much of the protein is produced by the novel-fused gene [1].
- Detection of gene fusions and other genomic rearrangements involves multiple methodologies, categorized by sequencing and non-sequencing approaches. Non-sequencing techniques, such as fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC), offer rapid turnaround time, cost efficiency, and allow morphologic confirmation of which cells contain the target, making them widely adopted in clinical practice. However, these methods have intrinsic limitations: FISH cannot reliably detect small intrachromosomal rearrangements or define fusion partners, and IHC provides only semi-quantitative data [5,6]. Reverse transcriptase polymerase chain reaction, though highly sensitive, depends on prior knowledge of fusion partners due to its reliance on gene-specific primers, limiting its utility for genes with numerous fusion variants [7]. NGS, encompassing both DNA- and RNA-based strategies, addresses many of these limitations by allowing simultaneous identification of multiple fusion events [8]. DNA-based NGS interrogates both exonic and intronic regions, but carries a risk of false negatives, necessitating confirmation with RNA-based assays, which focus specifically on expressed transcripts [9]. RNA-based NGS, particularly through targeted fusion panels, has become an essential complementary technique due to its high sensitivity for fusion transcripts [10,11]. Liquid biopsy–based detection, primarily via circulating tumor DNA analyzed by DNA-based NGS, is promising but also challenged due to DNA fragmentation, low concentrations, and technical artifacts that reduce sensitivity compared to tissue analyses [12,13]. Given these complexities, optimal diagnostic accuracy often requires the integration of orthogonal sequencing methods—using DNA and RNA sequencing in parallel—to achieve comprehensive fusion detection [14]. When a novel gene fusion is identified, functional characterization is necessary to assess its clinical significance. A recently introduced functional genomic strategy systematically evaluates the cellular impact of gene fusions, incorporating an integrated, evidence-based classification framework to prioritize their relevance [15]. Importantly, a recent retrospective analysis demonstrated improved clinical outcomes among patients harboring actionable gene fusions who received fusion-targeted therapies (n = 25) compared with those treated with just systemic therapies not matched to their fusions (n = 42) [16]. These findings underscore the clinical value of fusion testing and highlight the therapeutic potential.
- Non-Spitz melanocytic nevi
- To date most of the gene fusions in benign non-Spitz melanocytic nevi, although rare, were reported in congenital nevi, known to harbor missense pathogenic variants in NRAS and BRAF and with a remaining 25% being unknown mutation status [17]. Gene fusions were identified in wild-type and mutant for NRAS/BRAF/KRAS nevi.
- In 2007, Dessars et al. [18] reported two cases of congenital melanocytic nevi with BRAF fusions: first was FCHSD1::BRAF and a second one where the partner to BRAF was not determined. Neither of two cases had a BRAF or NRAS mutation [18]. In 2019, Mir et al. [19] reported potentially actionable AKAP9::BRAF fusion in a giant congenital melanocytic nevus subsequently successfully targeted by trametinib [19]. In 2022, Molho-Pessach et al. [20] reported a CUX1::BRAF fusion in a case of another giant congenital melanocytic nevus. A ZNF777::BRAF was fusion identified in a congenital nevus from a patient with multiple melanomas where one of the prior patient’s melanomas demonstrated the same fusion along with a TERT promoter mutation and MET copy gain [21]. In a cohort of 19 cases with wild type for NRAS and BRAF congenital nevi, gene fusions were identified in 12 cases (7% of the total 169 patient cohort), where BRAF fusions significantly outperformed RAF1 fusions. Ten different partner genes were identified (AGAP3, AKAP9, EEA1, GOLGA4, LCA5, MIER3, PHIP, QKI, SEC31A, and STRN3), where only EEA1 and GOLGA4 were recurrent partners. Clinically BRAF/RAF1 fusions are significantly associated with a hyperproliferative phenotype of the nevi (p < .001), chronic intractable pruritus, requirement for surgical intervention for tumor overgrowth and high sensitivity to MEK inhibitors [22]. Histopathology of BRAF-fusion driven non-Spitz nevi demonstrated variable patterns of desmoplasia and fibrosis. Two of the mentioned cases of congenital nevi harboring EVI5::BRAF and QKI::BRAF fusions were successfully treated with MEK inhibitors [22]. The study from Botton et al. [23] revealed another BRAF fusion partner, ZKSCAN5 (a zinc finger protein), in an intradermal melanocytic lesion from the buttock. Agrawal and Guo [24] reported another dermal melanocytic lesion with congenital features and bland nevoid cytomorphology also from a buttock of 44-year-old female with an EPS15:BRAF fusion. Recently Roy et al. [25] reported a case series of five giant congenital melanocytic nevi with BRAF fusions (two cases with AKAP9::BRAF, ATAD2::BRAF, ST13::BRAF, and TRIM4::BRAF) demonstrating the same hyperproliferative phenotype, pruritus, multiple satellite nevi, and desmoplastic stroma. Interestingly, one patient with ATAD2::BRAF fusion developed neurocutaneous melanosis. El-Rayes et al. [26] reported another case of congenital melanocytic nevus with SH2B1::BRAF fusion and associated neurocristic cutaneous hamartoma.
- In 2019, Baltres et al. [27] reported a case of rapidly growing mass from the lumbosacral giant congenital nevus, showing melanoma with myxoid and rhabdomyosarcomatous transformation, and harboring an in-frame SASS6::RAF1 fusion by RNA-based sequencing in both the melanoma and nevus components. Neither component had NRAS or BRAF mutations by sequencing. A RAF1 break-apart FISH assay revealed a balanced rearrangement in the nevus and an unbalanced one in the malignant component [27]. In 2019, Martins da Silva et al. [28] in study of twenty-one congenital melanocytic nevi reported two novel gene fusions in NRAS-wild type nevi - ZEB2::ALK and SOX5::RAF1 and one GGNBP2::MYO19 in NRAS-mutant nevus. The ZEB2::ALK and SOX5::RAF1 fusions were identified only in nevi and not in the unaffected skin, supporting the oncogenic potential of those two fusions. On the other hand, the GGNBP2::MYO19 fusion was also detected in the unaffected skin and was considered non-actionable [28]. Vinyals et al. [29] found the same SOX5::RAF1 fusion in a NRAS/BRAF-wild type congenital nevus. That study revealed that SOX5::RAF1 fusion transcript expression led to MAPK activation through increased levels of phosphorylated ERK protein in the cytosol of transduced cells; induced growth factor-independent cell growth in murine hematopoietic Ba/F3 cells and Melan-A immortalized melanocytes; and promoted tumor growth and lung metastasis in mice. It was suggested that SOX5::RAF1 fusion is an oncogenic functional fusion with heightened possibility to malignant transformation and potentially targetable with RAF1 kinase inhibitors or those targeting MAPK pathways [29].
- These studies show that BRAF and RAF1 kinase fusions represent recurrent genetic alterations in congenital and non-Spitz melanocytic nevi that usually lack mutations in BRAF, KRAS, or NRAS. These fusion events activate the MAPK pathway, frequently demonstrate fibrotic background on histopathology, should undergo close follow up and may be amenable to targeted therapy with RAF or MEK inhibitors.
- In 2021, the group of Houlier et al. [30] reported five cases harboring RASGRF2 fusions, in particular ATP2B4::RASGRF2 and ERBIN::RASGRF2 fusions, with a variety of clinical presentations ranging from large congenital nevus to melanoma ex-nevus as well as intermediate grade melanocytic lesions (melanocytomas). Most cases were present at birth, including a large congenital nevus, supporting a postzygotic origin of the RASGRF2 fusion, similar to BRAF and RAF1 gene fusions in nevi. Interestingly, in the reported malignant transformed case of that series, the melanoma demonstrated a peculiar histomorphology of mainly dermal expansion of dense sheets of epithelioid atypical melanocytes. In addition, this malignant case demonstrated BAP1 inactivation and authors hypothesized that RASGRF2 fusion induced the overexpression of a chimeric RasGRF2 protein leading to an oncogenic activation of the MAPK pathway, considering that all cases demonstrated RasGRF2 overexpression [30]. The RASGRF2 expression was reported in multiple other carcinomas including rapidly growing triple-negative breast cancer [31], invasive colorectal cancers [32] and lung carcinomas associated with poor prognosis [33]. The fusion partner ATP2B4, that was found three out of five cases, was also repeatedly reported as ATP2B4::PRKCA in pigmented epithelioid melanocytomas including congenital cases [34,35]. No gene fusions involving another gene partner ERBIN (ERBB2 interacting protein) have been previously reported in melanocytic lesions. Overall, melanocytic lesions harboring RASGRF2 fusions display unusual dermal morphological features and can progress to malignancy although more data is needed to prove it as an oncogenic driver.
- Other
- Perron et al. [36] reported four cases of melanocytic myxoid spindle cell tumor with ALK rearrangement (MMySTAR) including FBXO28::ALK, NPAS2::ALK, TPM3::ALK, and PPFIBP1::ALK. These lesions lose at least partially Melan-A expression and were interpreted as a potentially new variant of compound nevi linked to a kinase fusion [36].
- Conventional non-Spitz melanoma
- Unlike driver somatic mutations, such as BRAF, KIT, NRAS, NF1, GNAQ, or GNA11, gene fusions are rare in conventional non-Spitz melanomas with recently reported data indicating that only 2% of melanomas harbor it. Among melanomas with gene fusions, BRAF fusions were the most common, present in 0.8% (6/750) of cases, followed by RAF1 fusions in 0.7% (5/750), and ALK fusions in 0.3% (2/750). Melanomas with gene fusions were typically triple wild-type, lacking somatic mutations in BRAF, NRAS, or KIT. A recent study from Moran et al. [37] reported that all BRAF-fused melanomas (100%; 5/5) also consistently harbored TERT promoter mutations. Our study showed that only 44% (4/9) of BRAF-fused melanomas had TERT promoter mutations [38].
- The AGK is the most frequent fusion partner of BRAF in melanomas [37,39,40]. Other less common fusion partners include AGAP3, AGT7, ARMC10, CDH3, CHCDH3, CCT8, CCDC91, GTF2IRD1, DIP2B, FMN1, GTF21, MKRN1, PAPSS1, RAD18, SEPT3, TRIM24, TAX1BP1, and MYO5A [37-45]. Menzies et al. [46] reported PPFIBP2::BRAF fusion on a superficial spreading melanoma metastatic to brain and a KIAA1549::BRAF fusion on a metastatic acral lentiginous melanoma, both included in a trametinib and pembrolizumab trial. Kim et al. [47] added other new BRAF gene partners in mucosal non-Spitz melanomas such as ZNF767, NFIC, TNEM178B, and DGKI. Perron et al. [48] reported two cases of deeply infiltrative melanomas with sclerosing/desmoplastic features and an AKAP9::BRAF fusion where one of the cases also had MDM2 amplification. We have recently reported a case of fatal melanoma with RNF11:BRAF fusion also containing a TERT promoter mutation and showing peculiar epithelioid and rhabdoid morphologies, occasional giant pleomorphic cells and multinucleation, progressed with multiple metastases and reported non-responsive to therapy [49]. We also presented another case of melanoma with rhabdoid cytomorphology, multinucleation and occasional Reed-Sternberg–like cells resembling alveolar soft part sarcoma, that harbored a SBF1::BRAF fusion and a TERT promoter mutation [50]. The histopathologic features of BRAF-fused conventional bona fide melanomas seem to differ from those of Spitz melanocytic neoplasms with BRAF fusions.
- The recent data have shown variability in treatment responses for these tumors, with BRAF fusion potentially indicating a poorer prognosis and/or more aggressive behavior [37,39,49,51].
- RAF1-fused conventional melanomas are extremely rare and comprise only 0.6% of cases [52,53]. Available data shows RAF1 gene fusion partners such as ANO10, CCDC85A, CDH3, GCC2, GOLGA4, EFNB1, EFCC1, FCGRT, FYCO1, LRCH3, MAD1L1, MAP4, NF2, RUFY1, and SLC4A7 [37,53-58]. The study of Williams et al. [52] added additional RAF1 5′ fusion partners: MAP4 (n = 3), CTNNA1 (n = 2), LRCH3 (n = 2), GOLGA4 (n = 2), CTDSPL (n = 2), and PRKAR2A (n = 2), where 62% (23/37) of cases had TERT promoter mutation and 60% (24/40) had CDKN2A activating mutation [52]. We have encountered a case on the ankle in a 24-year-old female, resistant to therapy and fatal, triple wild-type melanoma with a MAP4::RAF1 fusion and TERT promoter mutation [59]. The study from Stransky et al. [60] reported 4 additional RAF1 gene fusion partners in conventional melanoma including LMNA, MPRIP, TRAK1, and CLCN6.
- Most of the reported RAF1-fused melanomas responded to targeted therapy and available in vitro studies also demonstrated sensitivity of RAF1 fusions to the multi-kinase inhibitor sorafenib and variably responsiveness to the MEK inhibitors [53,55,61,62].
- The study from Lezcano et al. [63] reported NTRK1 fusion partners including TRIM63, DDR2 and GON4L, and one NTRK2::TRAF2, where all lesions were immunoreactive for pan-TRK.
- Couts et al. [64] reported a mucosal melanoma case with an EML4::ALK fusion that was sensitive to ALK inhibitors. The study from Moran et al. [37] also reported two melanomas with EML4::ALK and MLPH::ALK. Perkins et al. [65] presented two cases of pediatric melanoma with ZEB2::ALK fusion arising within a congenital melanocytic nevus. There is a recent case of BAP1-inactivated melanoma associated with ALK fusion with unknown partner [66].
- The novel FGFR3::TACC3 fusion was reported by Lee et al. [67] in a case of conventional melanoma with plasmacytoid features from the orbit.
- Beyond Spitz lineage, MAP3K8 rearrangements occur in ~1.5%–1.7% of conventional melanomas in The Cancer Genome Atlas/other datasets, but most are C-terminal truncations, intronic region, rather than named fusions. MAP3K8-rearranged melanomas tend to have a low mutational burden and significant decrease of typical ultraviolet (UV)-mutational patterns, with frequency of UV-associated mutations (C>T and G>A) in comparison to MAP3K8 wild melanoma of 40% vs. 74% (p = .0023) [68]. The break-apart FISH strategy and sequencing-based approaches have the potential to identify patients with MAP3K8 truncating rearrangements. There are data of acral melanomas with MAP3K8:DEK fusion [69]. There is a recent case of a fatal mucosal melanoma with focal spitzoid features and MAP3K8::PAK2 fusion, CRKL amplification and hemizygous TP53 R280T mutation [70]. Although immune checkpoint therapy combinations are unlikely to benefit patients with melanomas carrying MAP3K8 truncations, these melanomas have displayed sensitivity to both MEK and ERK inhibitors, providing rationale for a dual combination approach [68].
- Similar to MAP3K8, the MAP2K1 gene in conventional, acral, and mucosal melanomas is commonly reported to be mutated or truncated rather than fused [45,71-73]. In the largest cohort of MAP2K1-mutated primary cutaneous melanocytic tumors. Ebbelaar et al. [73] reported these alterations occur across the melanocytic spectrum (common and congenital nevi, melanocytomas, spitzoid tumors, desmoplastic melanomas) and can be grouped in three classes: I (RAF-dependent), II (RAF-regulated), and III (RAF-independent). Class I appeared almost exclusively in melanomas with co-driver BRAF or NRAS alterations and correlated with aggressive behavior. Classes II and III often acted as sole drivers across nevi, melanocytomas, and melanomas and carried substantial metastatic risk—especially when associated with TERT-promoter mutations [73].
- Other infrequent fusions included CANT1::ETV4, CCDC6::RET, CLPTM1L:ADCY2, NOTCH1::GNB1, ADCY2::TERT, NAGS::MAST2, MDM2::GNS, MDM2::CCT2, PTEN::RPL11, TERT:ADCY2, TERT:PDCD1LG2, and PAK2::LOC646214 [37,69,74]. A recent study of acral melanomas reported another cohort of recurrently fused kinase genes included TRIO (11 fusions in 3 tumors), PAK1 (9 fusions in 6 tumors), DGKB (7 fusions in 4 tumors), and DCLK1 (3 fusions in 2 tumors) [45].
- We also recently added two cases of BCR-fused melanoma BCR::ZNF711 (zinc finger protein 711) and BCR::CYLC2 (cylicin 2), where mutations in BRAF, KIT, or NRAS, both cases underwent immunotherapy and remained without evidence of disease although functional significance of BCR fusions in melanomas remains unclear at this point [75].
- Spitz neoplasms
- Historically Spitz neoplasms represent a diagnostic “gray zone” in dermatopathology mainly due to low inter-observer agreement in terminology—such as 'true Spitz lineage' versus 'Spitz-like'—and inconsistent classification across a spectrum that includes benign Spitz nevi, lesions of uncertain malignant potential (atypical Spitz tumors or melanocytomas), and Spitz melanomas. The diagnostic complexity is further complicated by their variable biological behavior and potential for locoregional lymph node involvement. Spitz neoplasms can be classified into four groups: (1) mutations (HRAS mutations [with or without 11p amplification] and 6q23 deletions); (2) receptor tyrosine kinase fusions (ALK, ROS1, NTRK1, NTRK3, RET, MET, MERTK, LCK, and FGFR1); (3) serine/threonine kinase fusions and mutations (BRAF, RAF1, ERBB4, MAP3K8, MAP3K3, and PRKDC); and (4) other rare genomic aberrations [76-78]. All these driver gene alterations so far are considered mutually exclusive. In the receptor tyrosine kinase fusions group, the 3′ end intact kinase domain is placed under the control of the promoter of 5′ end partner gene followed by generation of a chimeric protein with functional kinase with constant overexpression of the RTK in melanocytes. The cellular localization of the chimeric protein—whether at the cytoplasmic membrane, within the cytoplasm, or in the nucleus—is determined by the biological properties of the 5′ fusion partner [79]. The serine threonine kinase fusions are less frequent and demonstrate similar mechanism to RTK fusions with BRAF partner while in MAP3K8 fusions, the 5′ partner retains the first eight exons of MAP3K8, with the fusion breakpoint occurring downstream. This results in the loss of the 3′ sequence encoding the regulatory tail that normally occludes the kinase domain [80]. The new oncogenic model was recently described with MAP2K1 micro-deletions and fusions involving RASGRF1 and RASGRF2 where the main driver is kinase modulator rather than the kinase itself [30,81,82].
- The current problem of Spitz neoplasms lies in the presence of clearly malignant tumors with spitzoid morphology and fatal outcomes, where growing data on oncogenic gene fusions may help segregate these “Spitz-like” tumors from “true Spitz lineage” and thus allow a different patient management. Truly malignant, fusion-driven Spitz neoplasms do occur but they remain relatively rare. In such cases, molecular alterations such as homozygous deletions of 9p21 (CDKN2A), 6p25 copy number gain, TP53 mutations or loss, PREX p.S658 L mutation, Rb gain or TERT promoter mutations may provide useful prognostic information and consideration of a diagnosis of Spitz melanoma [77,83-85]. Further understanding of gene fusions in Spitz neoplasms may aid in their diagnostic classification, risk stratification, and therapeutic decision-making.
- Up to 20% of Spitz nevi harbor copy number gains and/or activating mutations in HRAS, located at chromosome 11p15.5 [86]. Spitz nevi with this genomic alteration often exhibit desmoplastic features and/or gain of 11p as the only detectable copy number aberrations (isolated gain of 11p); however, this phenotype is not consistently present in all cases [87].
- Tyrosine kinase fusion–associated Spitz neoplasms
- ALK gene rearrangements have been identified in approximately 10%–20% of Spitz nevi, 15% of atypical Spitz tumors, and about 3% of Spitz melanomas [79,88,89]. The most frequent fusion partners include TPM3 and DCTN1, followed by NPM1, TPR, CLIP1, GTF3C2, MLPH, EEF2, MYO5A, EHBP1, and KANK1 [88,90-93]. The chimeric fusion protein upregulates MAPK and PI3K/AKT/mTOR pathway and can be inhibited by ALK inhibitors [79]. ALK-rearranged spitzoid tumors are well characterized in the literature: they occur in a wide age range, usually involve lower limbs and usually clinically are amelanotic and polypoid [90,94]. These ALK-rearranged Spitz melanocytic neoplasms demonstrate distinct histopathology: exophytic or polypoid tumors with fusiform-to-epithelioid melanocytes arranged in a nested, fascicular, or plexiform growth pattern with wedge-shaped base architecture and infiltrative border at the periphery, or rarely a bulbous to nodular ‘dumbbell’ growth pattern [90]. Angiomatoid histomorphology is also seen in ALK fusions [80]. Several authors reported a total 11 cases of Spitz neoplasms harboring MLPH::ALK and diagnosed them as Spitz nevi or atypical Spitz tumors (ASTs) with only one case of Spitz melanoma [91,92,95,96]. MLPH::ALK-driven Spitz lesions tend to occur in younger population (<40 years old) with slight female predominance, manifest as an exophytic or polypoid nodule characterized by a proliferation of fusiform-to-epithelioid melanocytes with consistent dermal mitotic activity and diffuse expression of cytoplasmic ALK by IHC except of one case [91,92,95,96]. We also encountered a case of MLPH::ALK-fused atypical Spitz tumor with perineural invasion. Interestingly, in case of EHBP1::ALK fusion the ALK IHC demonstrated a peculiar membranous pattern of staining. It is important to remember that although ALK IHC is available along with ROS1 and pan-TRK, the positive result is only a reflection of overexpression and is not confirmative for fusion [97]. The type of labeling (membranous, cytoplasmic, or nuclear) is also should not be considered diagnostic Spitz nevi vs AST vs melanoma since it is fully dependable to the 5′ end fusion partner.
- There are only a few documented cases of Spitz melanoma featuring ALK fusions [79,91,98]. Spitz melanoma with SLC20A1::ALK fusion, negative for BRAF, KIT, NRAS, TERT but harboring GRM3 that is known to upregulate the MAPK pathway [99]. Frederico et al. [100] reported a C2orf42::ALK fusion Spitz melanoma in a 5-year-old girl with lymph node metastasis as well as diffuse membranous staining for ALK. Raghavan et al. [98] reported two Spitz melanomas, both with fascicular cytomorphology, one with a DCTN1::ALK fusion and second with a fusion of the 3′ portion of ALK to an intergenic region of chromosome 11q. This resulted in a gain of the distal portion of ALK encoding the kinase domain, suggesting that a complex rearrangement resulted in an ALK kinase fusion as the oncogene in this case [98]. Neither gene fusion is diagnostic for Spitz melanoma although fusions involving ALK and MAP3K8 are among the most common rearrangements associated with progression to Spitz melanoma [97,101]. TERT promoter mutations rather than other factors such as deletions of the CDKN2A gene are highly predictive of an aggressive clinical course characterized by the development of distant metastases and a fatal outcome [101].
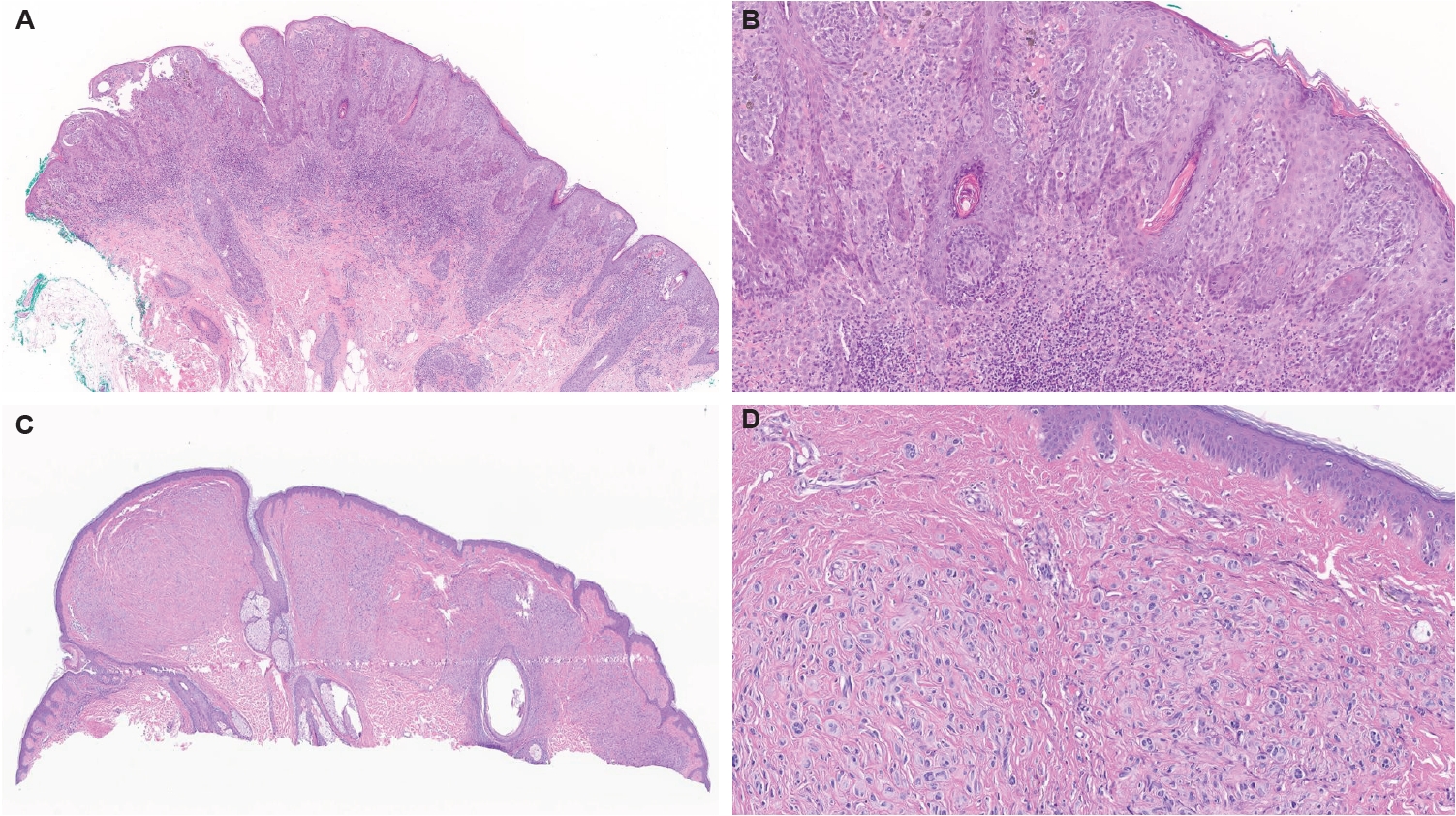
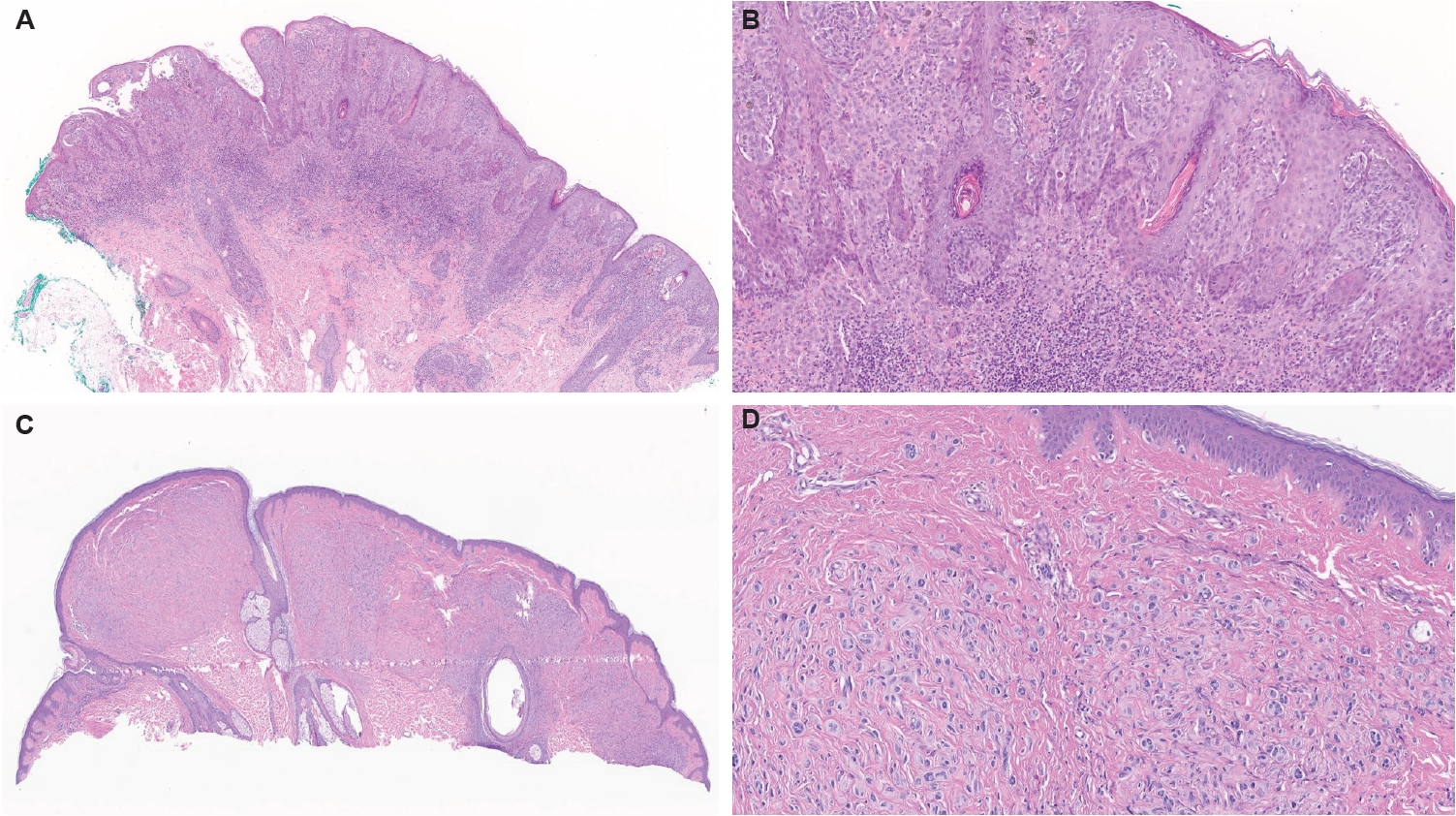
- Wiesner et al. [79] reported ROS1 fusions in 17% of Spitz neoplasms where 25.3% from detected fusions comprised benign Spitz nevi, 6.3% AST/melanocytomas, and 9% Spitz melanomas [85,102]. Therefore, ROS1 fusions in Spitz neoplasms have a tendency for favorable outcome [102,103]. Furthermore, only few reported cases of ROS1 fused Spitz melanomas reported to date, none of which resulted in distant metastases or death from disease [79,104]. ROS1 fusions have been reported to have several partner genes, including most prevalent PWWP2A (37% of cases) and TPM3 (31% of cases), followed by PPFIBP1, CAPRIN1, MYO5A, CLIP1, ERC1, FIP1L1, HLA-A, KIAA1598, MYH9, ZCCHC8, and GOPC [79,98,103,105,106]. The chimeric fusion product demonstrates constitutively increased phosphorylation activity, with increased activation of downstream MAPK and PI3K/AKT/mTOR signaling cascades [4,79]. We also encountered a classic Spitz nevus with PPFIBP1::ROS1-fused atypical Spitz tumor (Fig. 1A, B). There are also reports of ROS1 fusions in desmoplastic Spitz nevi [79,102], pigmented spindle cell nevi of Reed and one eruptive Spitz nevus [107].
- The clinical presentation of ROS1-fused Spitz lesions was well described by Gerami et al. [102]: age range of 3 to 58 years (mean of 19 years), no particular anatomical site predisposition, presented as pink to red papules. Although ROS1-fused Spitz neoplasms generally lack specific histopathologic characteristics in comparison to ALK-fused lesions, lesions with ROS1 fusions displayed a well-circumscribed epidermal and dermal proliferation of epithelioid and spindled melanocytes with mild to moderate atypia, low mitotic activity (~1.3/mm²) and Kamino bodies [102]. There was also transepidermal elimination of melanocytic nests and myxoid/mucinous stroma [103]. Immunohistochemically, the anti-ROS1 monoclonal antibody is quite specific but at the same time is typically weak; therefore it is recommended to confirm ROS1 rearrangement by FISH, polymerase chain reaction, or NGS [77]. Various immunohistochemical staining patterns have been reported, including diffuse granular cytoplasmic, dot-like, and nuclear expression; however, no consistent correlation has been established between these patterns and the subcellular localization of the different ROS1 fusion variants identified to date [108]. The implementation of NGS recently yielded a novel and potentially actionable ROS1 p.S1986P mutation, which has not been previously reported in Spitz nevus [85]. The group of Delsupehe et al. [78] reported two new fusions: LIMA1::ROS1, in a 17-year-old male with hypertrophic buccal lesion, histologically composed of epithelioid and fusiform melanocytes, that was subsequently classified as Spitz nevus; and LRRFIP2::ROS1, in a 73-year-old female with rapidly growing lesion on the ankle, composed of conspicuous epithelial component of single melanocytes with architectural disarray and epithelioid dermal component, later categorized as AST/Spitz melanocytoma. As was mentioned before, ROS1 fusions in melanoma and “favor melanoma” lesions are rare and include: GOPC::ROS1 in acral lentiginous conventional melanoma [109] and a case of unresectable Spitz tumor [105] with dramatic response to ROS1 targeting tyrosine kinase inhibitors—entrectinib and crizotinib in both cases; and a ROS1 fusions were also reported in 9.1% of spitzoid melanomas [79]; as well as ROS1 rearrangement was reported in a superficial spreading conventional melanoma but was likely nonfunctional [4].
- The NTRK1, NTRK2, and NTRK3 genes encode TrkA, TrkB, and TrkC receptors, which mediate melanocyte responses to neurotrophic signals [110,111]. NTRK gene fusions lead to Trk overexpression or activation, triggering MAPK, PI3K, and PLCγ pathways [112]. The frequency of NTRK fusions in Spitz neoplasms is approximately 10%–17% with NTRK1 being most prevalent followed by NTRK3 [113]. It is important to mention that pan-TRK and NTRK1 antibodies are widely used and cost-effective for screening for NTRK fusion, including available clones A7H66R and EPR173441 [114-118]. Pan-TRK immunostaining patterns differ by NTRK fusion type, with NTRK1 fusions typically showing strong cytoplasmic staining and NTRK3 fusions presenting as weak, granular, or nuclear-cytoplasmic staining. Molecular analyses (e.g., FISH, RNA sequencing) are necessary to confirm the specific fusion partner [97].
- NTRK1 encodes the TRKA receptor, a key oncogenic driver and promising target for cancer therapy due to its pivotal role in tumorigenesis. NTRK1 activation triggers cell signaling through pathways such as PI3K/AKT and MAPK [79]. NTRK1 fusions have been found across the entire Spitz spectrum including Spitz nevi, AST/melanocytoma, and Spitz melanomas [79,114]. Most reported partner genes include LMNA, PRDX1, TPM3, TP53, and KHDRBS1 [119] where LMNA was predominantly reported in Spitz nevi followed by atypical Spitz tumors and some spitzoid melanomas that harbor additional findings such homozygous CDKN2A deletion without known recurrence [111]; PRDX1 was reported in atypical Spitz tumor [120]; TPM3 in atypical Spitz tumor with associated heterozygous loss of CDKN2A and recurrence; TP53 and KHDRBS1 both were also reported in atypical Spitz tumors [111]. Histopathologically, NTRK1 fusion–positive tumors often display classic spitzoid morphology, with rare cases reported to show some rosettes, filigree-like epidermal rete ridges, lobulated nests or exhibit prominent maturation [121], though no specific histopathologic features reliably predict their presence. Unlike ALK-rearranged neoplasms, which more commonly exhibit plexiform or fascicular growth, NTRK1-positive lesions more frequently contain epithelioid-to-spindled cell cytomorphology [117].
- NTRK1 fusions were also reported in non-Spitz melanomas: NTRK1::TRIM63 in nodular melanoma, NTRK1::DDR2 in nodular melanoma from umbilical region with metastasis to colon, and a NTRK1::GON4L in nodular melanoma from back with metastasis in the duodenum co-occurred with NRAS Q61L mutation [63]. Lee et al. [122] reported NTRK1 translocation in another spitzoid melanoma which co-occurred with homozygous deletion of chromosomal 9p21 by FISH with positive lymph node and no recurrence. Raghavan et al. [98] reported another melanoma case with TPM3::NTRK1 fusion along with gain of chromosome 17 and loss of chromosome 5p upstream of TERT which possibly altered TERT promoter or enhancer sequences.
- While NTRK2 fusions are relatively rare compared to other fusions like NTRK1 and NTRK3, their identification in melanocytic lesions is important due to the possibility of targeted cancer therapies. The NTRK2::TFG fusion was reported in Spitz nevus which histopathology reminded pigmented spindle cell nevus of Reed with diffuse pan-TRK cytoplasmic immunoreactivity and negative for ALK, ROS1, and BRAFV600E [123]. Mansour et al. [124] reported a series of five Spitz tumors with SQSTM1::NTRK2 where all patients were female, all cases demonstrated diffuse pan-TRK staining and peculiar, prominent dendritic processes, highlighted with Melan-A immunohistochemistry, as well as conspicuous solitary or clustered hyaline. The junctional component showed horizontally oriented junctional nests and conspicuous lentiginous, single-cell growth in the follicular epithelium [124]. A NTRK2::TRAF2 fusion was also reported in a primary spitzoid melanoma, superficial spreading type, arising from perianal skin [63].
- NTRK3 fusions can be detected in 0.7% of Spitz tumors. They are observed in pigmented spindle cell nevus of Reed and in a subset of Spitz nevi with schwannomatous morphology with MYO5A being the most frequent partner, commonly affecting children [125]. Wang et al. [126] also reported this frequent occurrence in younger-age patients. NTRK3 gene fusions were reported by Yeh et al. [121] with fusion partners including ETV6, MYO5A, and MYH9, with a median age of 10 years. de la Fouchardiere et al. [127] described different features depending on the gene partner. NTRK3::ETV6 occurred in younger patients and had epithelioid or classic Spitz cytomorphology while NTRK3::MYO5A lesions contained spindled cells arranged in fascicles with neuroid features such as pseudo-Verocay bodies. They detected a chimeric product from NTRK3::ETV6 localization in the nucleus and cytoplasm, followed by intense nuclear and less intense cytoplasmic pan-TRK immunohistochemistry; and localization of NTRK3::MYO5A chimeric product in dendrites, followed by linear pan-TRK immunopositivity [127]. The MYH9::NTRK3-fused tumors are composed of syncytial epithelioid cells, with central desmoplastic stroma and peripheral collagen trapping [115,127]. The most recent NTRK3 gene fusion partner was reported by Della Mura et al. [128] as VIM::NTRK3 in a 10-year-old girl in atypical Spitz tumor/Spitz melanocytoma showing peculiar granular cytoplasmic and perinuclear dot-like pattern on pan-TRK IHC. While occasional involvement of regional lymph nodes has been observed, distant metastases and poor prognoses have not been documented to date.
- About 3%–4% of Spitz neoplasms were found to have RET fusions with CCDC6, KIF5B, LMNA, GOLGA5, and MYO5A [37,79,129,130]. Additionally, Donati et al. [129] reported other partners: OPTN in three cases, and AGAP3, NCOA4, ERC1, and MYH9. These RET-fused Spitz neoplasms do not demonstrate a definitive characteristic tumor cytomorphology, although nevoid cytomorphology may be associated with KIF5B, neuroid-like with MYO5A, epithelioid with LMNA and CCDC6 and transepidermal elimination/floating intraepithelial nests of pigmented melanocytes with OPTN [129]. Kim et al. [130] reported a case series where all RET-fused Spitz neoplasms demonstrated similar morphological features, i.e. plaque-like silhouette with expansile and dyshesive nests of monotonous predominantly epithelioid melanocytes without high grade atypia or pleomorphism. Although these RET fusions have been identified in all three Spitz nevi, atypical Spitz tumors, and Spitzoid melanomas, the available follow-up data indicate a favorable prognosis. The therapeutic potential for vandetanib and cabozantinib in these lesions is under investigation [131].
- MET-fused Spitz neoplasms have been rarely reported, harboring a diverse range of fusion partners, including TRIM4, ZKSCAN1, LRRFIP1, PPFIBP1, EPS15, and DCTN1 [85,88,132,133]. The chimeric fusion product activates the MAPK, PI3K/AKT, and PLCg1 pathways [88]. The most recent study from Roy et al. [134] discovered a significant addition to the novel MET gene fusion partners cohort: CPSF7, DCTN1, KIF5B, KLHL7, LRRFIP1, TRIM4, and ZKSCAN1, being diagnosed in low grade melanocytomas or nevi, and EPS15, MLPH, PPFIBP1, and TRIM4 in high grade melanocytomas or melanomas [134]. This study identified three main histopathologic patterns in MET-fused melanocytic neoplasms: “plexiform maturing fascicles”, “Reed-like” pattern, and “BAP1-deficient-like” pattern [134]. All cases with available follow-up have demonstrated indolent clinical behavior.
- Serine-threonine kinase-rearranged Spitz tumors
- BRAF-fused Spitz neoplasms along with MAP3K8 fusions are among the fusion driven Spitz subtypes that are likely to be diagnosed as Spitz melanoma. Activating point mutations of BRAF are not characteristic for Spitz neoplasm while BRAF fusions are relatively common with overall prevalence of 5%–5.7% with 14% reported as Spitz nevi, 45% as AST, and 41% as Spitz melanoma [3,79,89,98,132,135,136]. BRAF, a proto-oncogene on chromosome 7q encoding a serine/threonine kinase, when fused, loses its autoinhibitory CR1 and CR2 domains, retaining only the active CR3 kinase domain, thereby driving MEK-ERK pathway activation [41].
- The most reported BRAF gene fusion partners include CLIP2, AKAP9, EML4, AGK, BAIAP2L1, CEP89, CUX1, DYNC1/2, LSM14A, MAD1L1, MLANA, MYO5A, MZT1, NRF1, SKAP2, SLC12A7, SOX6, TRIM24, NUDCD3, PLIN3, KCTD7, and ZKSCAN5 [23,43,48,79,98,135-138]. The AGK and AKAP9 are the most frequent partners. Roy et al. [139] reported two desmoplastic Spitz nevi with TMEM106B::BRAF in one patient with a ring chromosome 7. Sorino et al. [140] reported an atypical Spitz tumor on the left outer ear in 13-year-old boy with TAX1BP1::BRAF, a finding that was previously reported only in non-Spitz melanomas. In the study by Sharma et al. [141] there were additional BRAF gene fusion partners: AST with TRIM33, TRIM24, RAB3GAP2, ZNF777, CDC42BPB, COBL, GOLGA4, KLC1, AGAP3, GTF2I, OSBPL1A, SND1, BIDC1, and Spitz melanoma with ERC1 [141]. Roy et al. [142] reported additional novel BRAF partners such as WDR91, ERC1, GTF2I, KIFAP3, RECQL, RNF11, RP2, and TMEM178B in “favor benign” Spitz neoplasms and EEA1, TRIM33, NRF1, ZKSCAN1 in “favor or concerning for melanoma” Spitz neoplasms.
- Clinically, the BRAF-fused Spitz lesions usually present as a pink papule on the extremities of young patients, predominantly females [136]. Histopathologically, in 2023, Roy et al. [142] reported a series of 58 cases with BRAF gene fusions and described three main histopathological presentations: (1) large epithelioid melanocytes arrayed as single cells embedded in a markedly fibrotic stroma (“buckshot” pattern); (2) polypoid growth with a whorled arrangement of cords and single melanocytes within a desmoplastic stroma (“cords in whorled fibrosis” pattern); and (3) fascicular growth of spindled melanocytes (“spindle-cell fascicles” pattern). These morphologic patterns did not distinguish benign and malignant melanocytic tumors with BRAF fusion [142]. In general, BRAF-fused Spitz neoplasms often lack well-organized nest formation and are primarily composed of large epithelioid cells with marked cytologic atypia and sclerotic stroma [89]. Marked fibrosis was more frequently present in BRAF-rearranged cases than in other fusions (p = 5 × 10-5) [116].
- In the large study of 38 cases and a meta-analysis performed by Sharma et al. [141], the BRAF-fused Spitz neoplasms lacked PRAME expression (24/24) and maintained p16 expression (16/21) with wild-type TERT promoter. Two of the cases developed lymph node metastasis although no recurrence or distant metastasis (mean follow-up time 26.1 months) [141]. Additional data for sentinel lymph node metastases were reported [104,135,142]. On the other hand, there is a case of 14-year-old boy with an EML4::BRAF fusion Spitz melanoma who developed lung, liver, and brain metastases while on ipilimumab and temozolomide and expired 18 months after diagnosis; the tumor also harbored a hotspot TPM [135]. There is another reported MAD1L1::BRAF fusion in fatal case of Spitz melanoma with TERT promoter mutation, CDKN2A/B deletions, MTAP deletion, MITF amplification, and PIK3CB amplification, who developed multiple distant metastasis [143].
- Donati et al. [144] reported two Spitz nevi and 1 Spitz melanoma with RAF1 rearrangement with the following gene fusion partners: CTDSPL and ATP2B4 in nevi cases and PPAP2B in melanoma case with associated homozygous deletion of 9p21 (CDKN2A) in greater than 80 % of enumerated cells. Serine–threonine kinase–rearranged Spitz tumors appear to acquire additional chromosomal aberrations more readily than receptor tyrosine kinase–rearranged Spitz neoplasms, with earlier malignant transformation that may be driven by CDKN2A alterations [47,145].
- The MAP3K8 is a serine-threonine kinase that directly phosphorylates MEK and activates ERK1/2. The truncating mutations and fusions of MAP3K8 eliminate the autoinhibitory C-terminal domain from the translated protein then leading to increased catalytic activity of the kinase and increased functioning of MEK1/2-ERK1/2 pathway, which is involved in cell proliferation, division, and differentiation [146]. Recent studies, using transcriptome sequencing, found structural initiating oncogenic alterations such as MAP2K1 in-frame deletions, MAP3K3 fusions, and MAP3K8 kinase fusions or truncations, in 9%–33% of Spitz neoplasms where non-BRAF MAPK gene alterations represented the most frequent driver in Spitz melanomas (22%) [147,148].
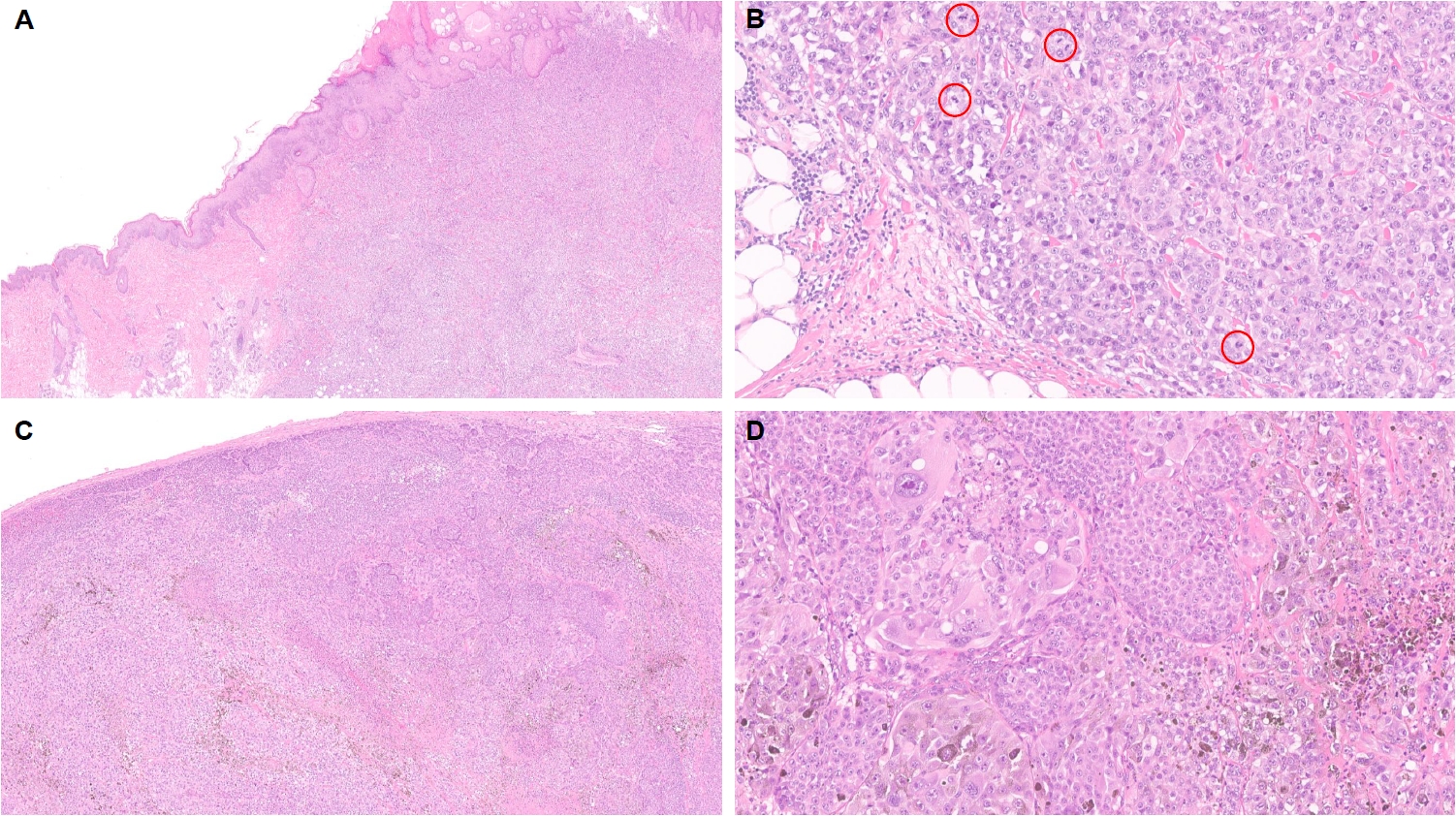
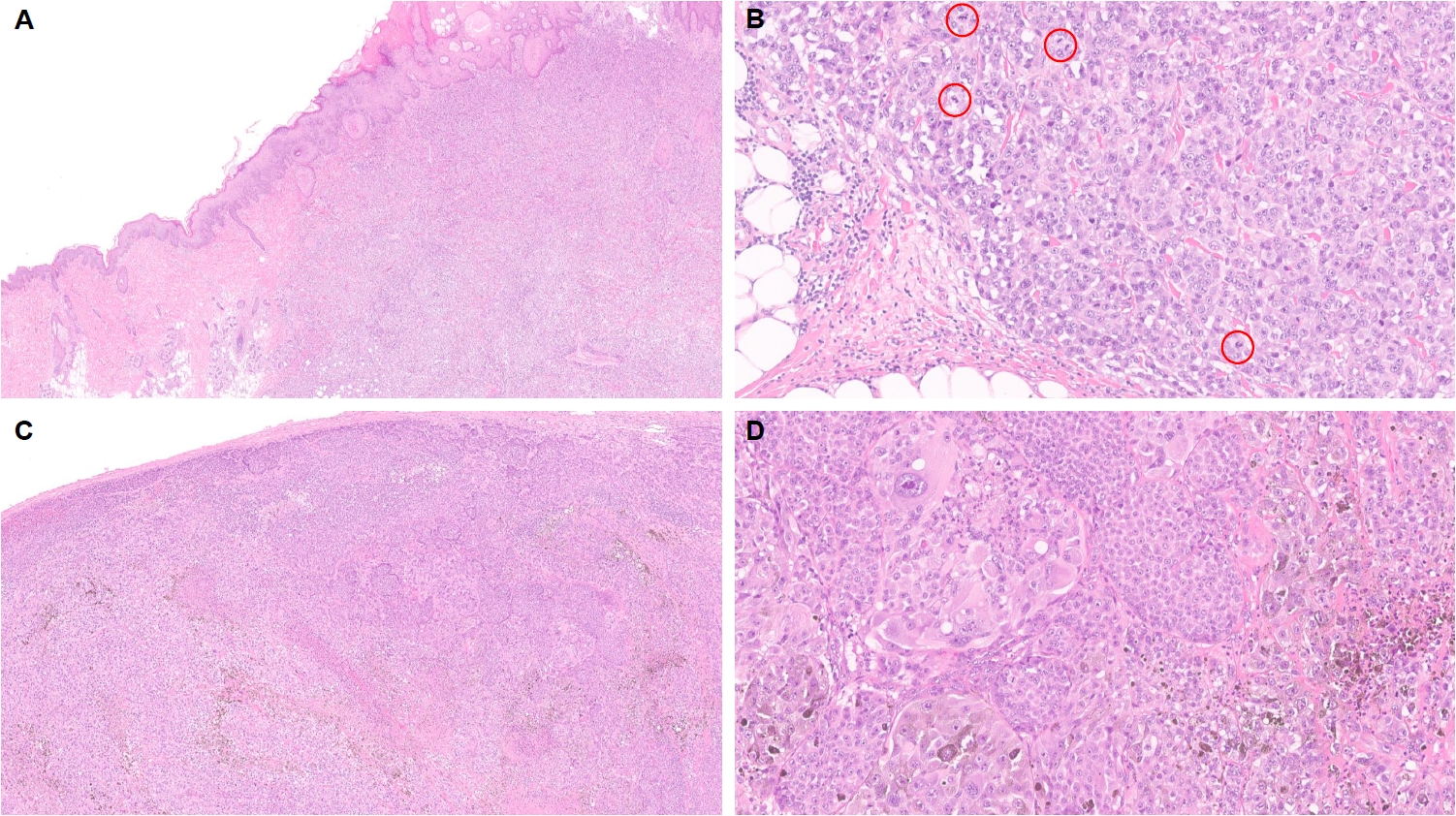
- Of the reported MAP3K8-fused Spitz neoplasms, 8% were classified as Spitz nevi, 40% as AST, and 52% as Spitz melanomas [98,145,148]. Among other serine–threonine kinase as well as tyrosine kinase fusions, the MAP3K8 fusion was the most common alteration observed in AST or Spitz melanoma (n = 25, 33%), while such alteration was quite rare in the group of Spitz nevi (n = 6, 2%) [116]. MAP3K8 gene fusion partners include ATP2A2, CCNY, CDC42EP3, CUBN, DIP2C, GNG2, LINC00703, LYZL2, MIR3681HG, PCDH7, PIP4K2A, PRKACB, RSU1, SFMBT2, SLC4A4, SPECC1, STX7, SVIL, UBL3, and ZFP36L1 [85,98,138,145,148]. Sibira et al. [149] also recently reported a case of Spitz melanoma with MAP3K8::ABLIM1 with associated GRIN2A, TP53, and TERT promoter mutations with rapid distant metastasis to lung. MAP3K8-fused Spitz neoplasms frequently harbor additional prognostically important genomic alterations such as biallelic inactivation or deletion of CDKN2A/B [85,98,116,145,148]. We encountered a case of MAP3K8-rearranged Spitz melanoma with unknown fusion partner, inactivating mutations involving CDKN2A and CDKN2B, and a TERT promoter mutation, with a Breslow thickness of 14.0 mm and lymph node metastasis (Fig. 2).
- Clinically, Spitz neoplasms harboring MAP3K8 fusions usually present as asymmetric, exophytic, pigmented lesions on the lower extremities, affecting patients of all ages with a slight female predominance [145,147]. Multiple other studies reported that MAP3K8-fused Spitz neoplasms presented as a dome-shaped or nodular lesion with overlying epidermal hyperplasia, and were associated with predominant epithelioid morphology, high-grade cytological atypia, multinucleated giant cells, and p16 loss [98,116,145,147,148].
- Although lymph node metastases are frequently reported, widespread metastatic disease is rare [145]. The data are limited but appeared to be a short-lived response to MEK inhibitor in a Spitz melanoma with a GNG2::MAP3K8 fusion [138].
- The proto-oncogene MAP2K1, situated at 15q and encoding a serine–threonine kinase integral to the RAF-MEK1/2-ERK1/2 signaling axis, exhibits recurrent mutations in Spitz neoplasms, mostly in-frame deletions involving exon 2 and 3, predominantly in atypical and malignant Spitz tumors which, similar to MAP3K8 fusion events, disrupt the autoinhibitory MEK domain and result in constitutive downstream pathway activation [81]. These mutations are fairly rare in Spitz neoplasms, with limited data available to date [116,148,150,151]. Whether MAP2K1-altered Spitz neoplasm harbor a distinctive histopathologic feature is questionable due to a high prevalence of additional genomic alterations [81]. Based on the available literature, MAP2K1-mutated Spitz neoplasms clinically present as small, pigmented, flat to slightly elevated lesions, predominantly on the lower extremities of young females. They tend to be wedge-shaped, compound or intradermal, with large epithelioid cells showing moderate to severe nuclear pleomorphism arranged in nests and showing a plexiform growth pattern, poor maturation and a tendency to converge around adnexal structures and neurovascular bundles [116,148,150,151]. In contrast to MAP3K8-fused Spitz neoplasms, these lesions often fail to demonstrate epidermal hyperplasia while frequently containing melanophages [81,150].
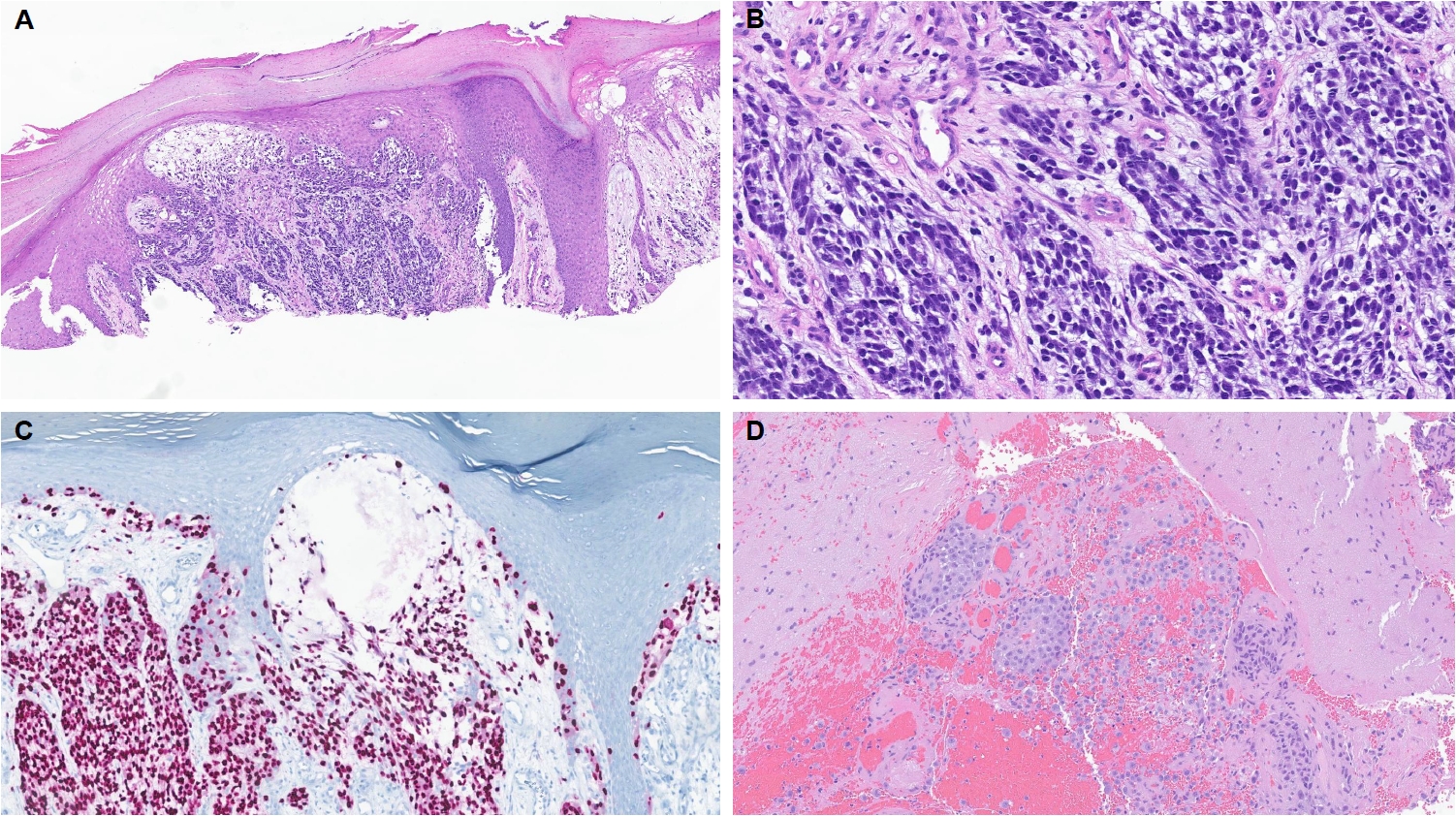
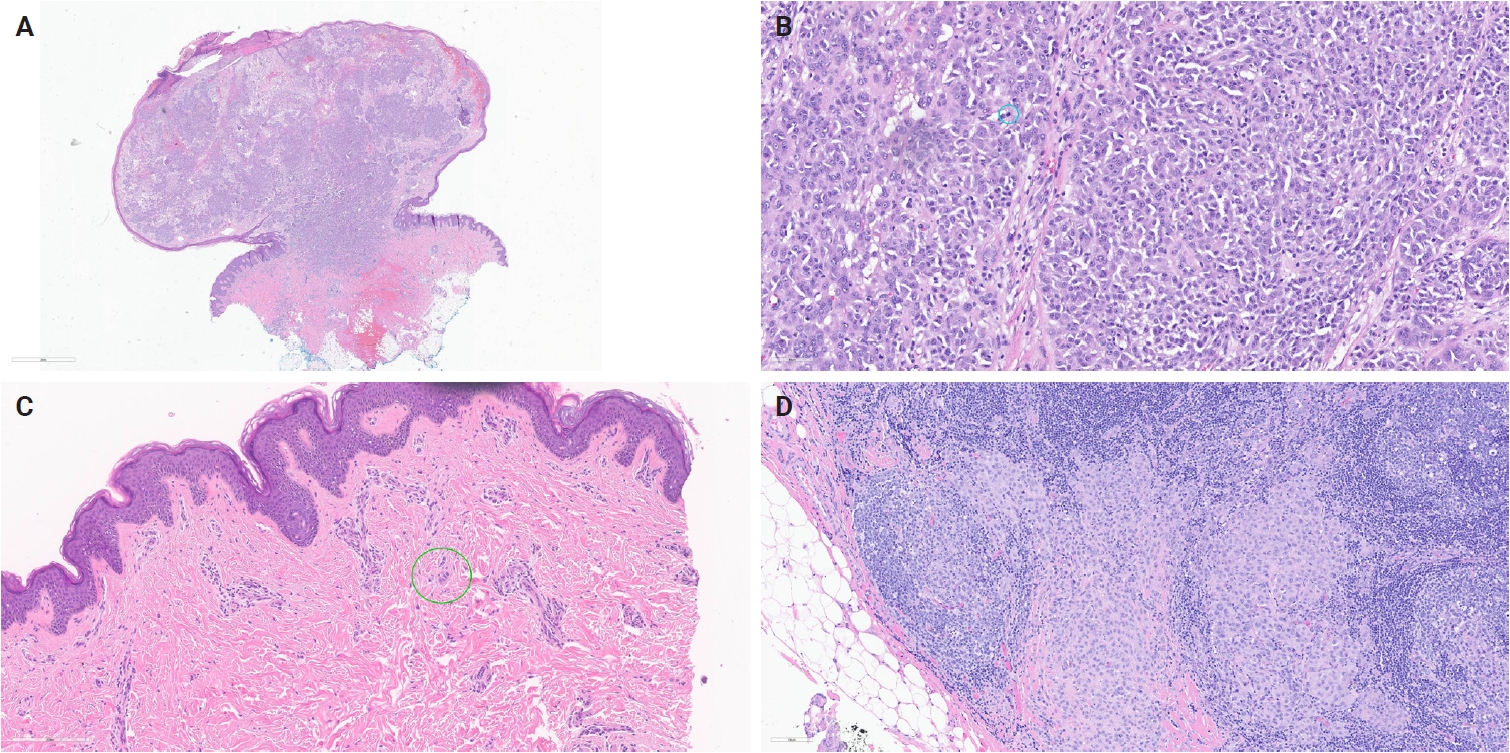
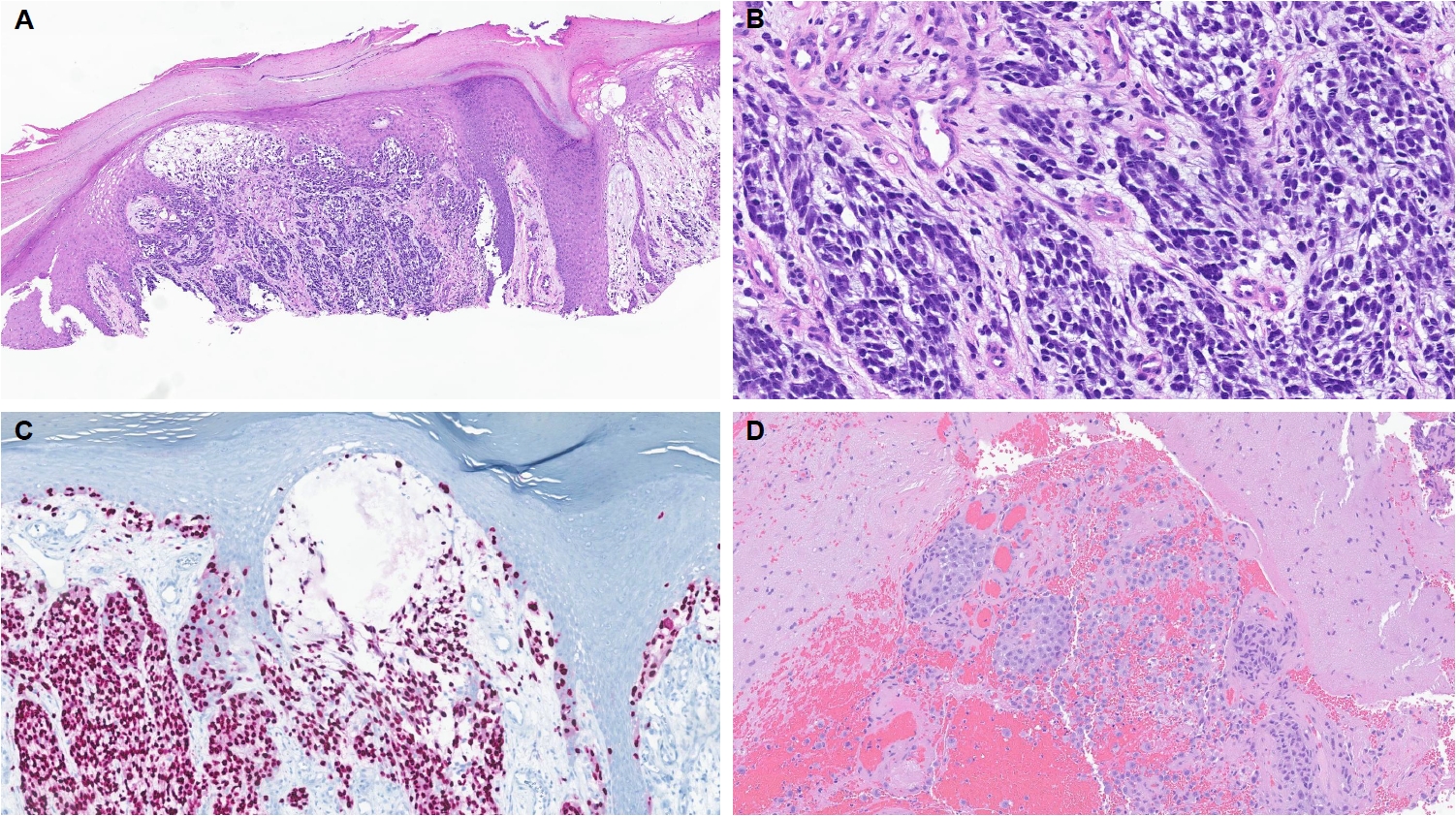
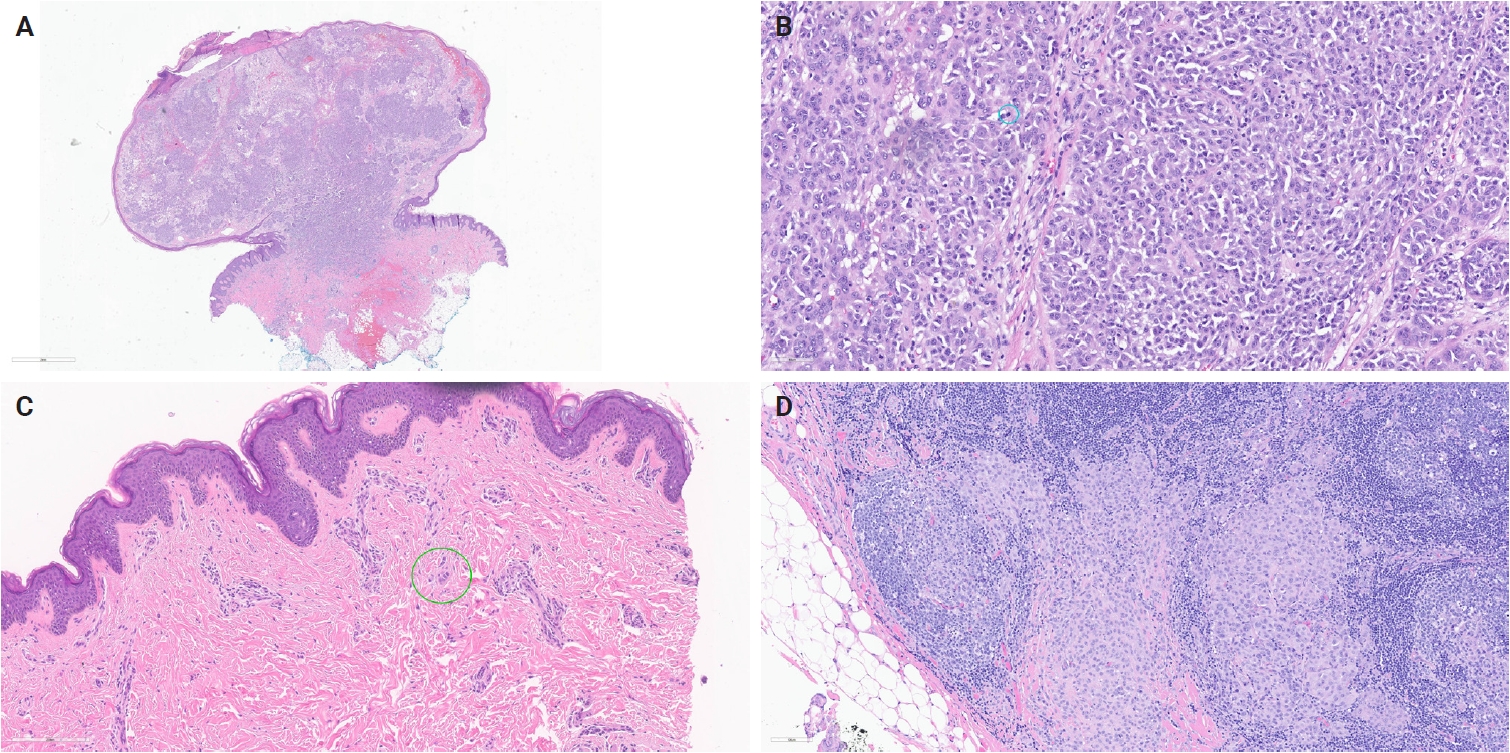
- Kervarrec et al. [152] reported an exophytic-shaped Spitz nevus without architectural or cytological atypia and a LMNA::MST1R fusion, where MST1R (macrophage stimulating 1 receptor) is a transmembrane tyrosine kinase receptor homologue of MET receptor, a known gene in Spitz tumors pathogenesis. The complex three-way translocation TRPM1::PUM::LCK inducing LCK mRNA overexpression was reported in case of agminated Spitz nevi occurring in a giant congenital, hyperpigmented macule without any other known translocation; authors suggested the possibility of additional “second hit” mutation beyond the initial gene fusion [153]. Quan et al. [85] reported another novel gene fusions, MYO5A::FGFR1, MYO5A::ERBB4, and PRKDC::CTNNB1, as well as a novel potentially oncogenic ROS1 p.S1986P mutation. We encountered another MYO5A::FGFR1-fused atypical Spitz tumor with desmoplastic stroma and prominent plump melanocytes (Fig. 1C, D). We recently also received a case of melanoma with Spitzoid features with locoregional metastasis and brain metastasis with a novel LOC107984974::RARA fusion (Fig. 3). Lastly, we want to report our new case of invasive melanoma with Spitzoid features and Breslow thickness of 8.8 mm from the right knee in 8-year-old girl, with ETV6::ZNF784 fusion, characterized by multiple microsatellitosis and lymph node metastases; the patient also had and a synchronous/or agminated compound melanocytic lesion with the same genetic alteration on the left fifth finger (Fig. 4).
- Pigmented spindle cell nevus of Reed
- VandenBoom et al. [125] analyzed twenty-three pigmented spindle cell nevi (PSCNs) and found that 78% harbored gene fusions being NTRK3 most frequent with ETV6 and MYO5A being the most common 5′ partners. Other fusions included MYO5A::MERTK (2), MYO5A::ROS1, MYO5A::RET, and ETV6::PITX3 [125]. Giubellino et al. [154] added two additional cases of PSCN of Reed with MYO5A::NTRK3 fusion. In 2021, Goto et al. [123] reported a TFG::NTRK2 fusion in PSCN. Those Spitz lesions with NTRK3 fusions develop in younger patients [121]. Predominantly on the extremities and show expansile nests of heavily pigmented spindle-shaped melanocytes with a plaque-like silhouette [125,126]. There is a recent PSCN in 61-year-old male with a SQSTM1::NTRK2 fusion and lacking alterations of ALK, ROS1, RET, NTRK1, and NTRK3 genes [155]. The observed BRAF V600E expression and older age may suggest the diagnosis of PSCN [156].
- Pigmented epithelioid melanocytoma
- Pigmented epithelioid melanocytoma (PEM) is an intermediate grade lesion with relatively frequent involvement of regional lymph nodes but rare distant metastasis [157,158]. There are two main molecular pathways for PEM: (1) fusions involving PRKCA, usually in young patients, with solid sheets of monomorphic, pigmented epithelioid melanocytes [34,35,159]; and (2) inactivating alterations in PRKAR1A with a preexisting mutation of BRAF or NRAS which demonstrate variable histomorphology and often demonstrate a conventional nevus component [3]. In addition, some lesions have alterations in GNA11 and GNAQ, changes commonly seen in blue-nevus-type neoplasm [159]. The reported PRKCA partners include ATP2B4, RNF13, SCARB1, CD63, MAP3K3, and ITGB5 [34,35,159,160]. The other rare gene fusions reported are MYO5A::NTRK3, TPR::NTRK1, NTRK3::SCAPER, CD63::PRKCB, and HTT::PKN1 [159,161-163].
- Blue nevi group melanocytic neoplasms
- PRKC-fusion melanocytic tumors have now been reclassified into the blue-nevus family with mostly benign course after excision and occasional cases of melanoma, often with BAP1 loss [164]. The study of 51 cases from de la Fouchardiere et al. [165] identified that PRKC-fused melanocytic neoplasms involve PRKCA in 35 cases, PRKCB in 15 cases, and PRKCG in one case. The reported gene fusions among PRKC-fused lesions include PTPRJ::PRKCB, ATP2B4::PRKCA, PTPRJ::PRKCA, SLC44A1::PRKCA, RNF13::PRKCA, LAMTOR1::PRKCA, HM13-PRKCA, EDNRB-PRKCA, SCARB1::PRKCA, PTTG1IP-PRKCB, EMP1-PRKCB, MLNA-PRKCG, and CD44-PRKCB [164,165]. PTPRJ and SLC11A2 were involved in both PRKCA and PRKCB fusions [165]. In two of the melanoma cases harboring PRKC fusions there was a BAP1 loss [60,165,166]. PRKC-fused cases are dermal‐based lesions composed of medium‐sized, epithelioid/oval cells with distinct hypercellular areas and a sheet‐like pattern alternating with areas with less compact oval/spindled melanocytes and fibrotic stroma, and overall minimal to light pigmentation [164,165].
- While activating mutations in GNAQ, GNA11, CYSLTR2, and PLCB4 genes are considered the main oncogenic drivers of blue nevi and blue malignant melanocytic tumors, the recent study reported an alternative oncogenic pathway involving GRM1 fusions such as MYO10::GRM1 and ZEB2::GRM1 fusions as well GRM1 rearrangement with an unknown partner identified by FISH [167]. Of note, both blue nevi-like melanomas demonstrate MYO10::GRM1 fusion with SF3B1 co-mutation.
- Deep penetrating melanocytic neoplasms/WNT-activated melanocytomas
- Yeh et al. [168] showed that deep penetrating melanocytic nevi/melanocytomas harbor activating mutations in the β-catenin and MAPK pathways, with a single reported AKAP9::BRAF fusion; progression to malignant counterparts appears to require additional alterations such as TERT-promoter mutations and CDKN2A loss.
- Other melanocytic neoplasms
- Gene fusions have also been reported as oncogenic drivers in other tumors that demonstrate melanocytic differentiation.
- The cutaneous melanocytic tumor with CRTC1::TRIM11 fusion, present as slowly growing, well‑circumscribed dermal nodule on adults across a broad age range and with no consistent anatomic distribution. It is composed of spindle and epithelioid melanocytes organized in nests, fascicles, or bundles and separated by delicate collagenous septa. IHC reveals consistent expression of S100, SOX10, and MiTF, with variable positivity for other melanocytic markers such as Melan‑A, PRAME, and HMB‑45, and nearly universal nuclear TRIM11 expression [169,170]. Initially reported with rare regional metastasis but recently reported to demonstrate more aggressive behavior [171-175].
- Subsequently, the three pediatric dermal tumors resembling CRTC1::TRIM11 tumor were described driven by a MED15::ATF1 [176]. They typically occur as dermal nodules on the head and neck (arm, cheek, scalp), some with epidermal involvement or ulceration, and may show early regional lymph nodes spread. They are highly cellular, composed of monomorphic oval-to-round epithelioid cells arranged in vague nests and short fascicles within a fibrotic stroma, with high mitotic activity (6–12/mm²). They demonstrate strong, diffuse nuclear SOX10 and MITF expression, variable S100 positivity, and focal reactivity for Melan-A, HMB-45, and pan-melanoma markers [176].
- There are two additional types of lesions driven by ACTIN::MITF (ACTB::MITF, ACTG1::MITF) and MITF::CREM. They also typically present as dermal nodules—often occurring on the extremities or head and neck—in predominantly adult female patients. They both exhibit clear cell features on histopathology and consistent MITF expression and melanocytic markers (such as HMB-45 antigen and Melan-A), variable S100 protein, and typically negative SOX10 in most ACTIN::MITF cases, whereas MITF::CREM tumors tend to mirror the phenotype of CRTC1::TRIM11 neoplasms with more frequent SOX10 and S100 expression [177-182].
ROLE OF GENE FUSIONS AND METHODS OF DETECTION
BRAF
RAF1
RASGRF2
BRAF
RAF1
NTRK
ALK
FGFR
MAPK
Other
HRAS
ALK
ROS1
NTRK 1/2/3
NTRK1
NTRK2
NTRK3
RET
MET
BRAF
RAF1
MAPK
MAP3K8
MAP2K1
Other gene fusions in Spitz neoplasms
- Gene fusions define and drive distinct, clinically meaningful subsets across all melanocytic tumors. Recognizing fusion-associated histomorphologies and using screening immunohistochemistry, if applicable, with confirmation by FISH or RNA-based sequencing improves diagnosis, classification, and possible selection for targeted therapy.
CONCLUSION
Supplementary Information
Ethics Statement
Not applicable.
Availability of Data and Material
All data generated or analyzed during the study are included in this published article (and its supplementary information files).
Code Availability
Not applicable.
Author Contributions
Conceptualization: VL, WCC. Data curation: VL. Formal analysis: VL. Investigation: VL, LE. Methodology: VL. Project administration: VL. Resources: VGP, WCC. Supervision: VL, WCC. Validation: VL. Visualization: VL. Writing—original draft: VL, LE. Writing—review & editing: VL, VGP, WCC. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
- 1. Salokas K, Weldatsadik RG, Varjosalo M. Human transcription factor and protein kinase gene fusions in human cancer. Sci Rep 2020; 10: 14169.ArticlePubMedPMCPDF
- 2. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017; 45: D777-83. ArticlePubMedPMC
- 3. Quan VL, Panah E, Zhang B, Shi K, Mohan LS, Gerami P. The role of gene fusions in melanocytic neoplasms. J Cutan Pathol 2019; 46: 878-87. ArticlePubMedPDF
- 4. Shalin SC. A review of kinase fusions in melanocytic tumors. Lab Invest 2017; 97: 158-65. ArticlePubMedPDF
- 5. Solomon JP, Hechtman JF. Detection of NTRK fusions: merits and limitations of current diagnostic platforms. Cancer Res 2019; 79: 3163-8. ArticlePubMedPMCPDF
- 6. Pecciarini L, Brunetto E, Grassini G, et al. Gene fusion detection in NSCLC routine clinical practice: targeted-NGS or FISH? Cells 2023; 12: 1135.ArticlePubMedPMC
- 7. Zito Marino F, Buono S, Montella M, et al. NTRK gene aberrations in triple-negative breast cancer: detection challenges using IHC, FISH, RT-PCR, and NGS. J Pathol Clin Res 2023; 9: 367-77. ArticlePubMedPMC
- 8. Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci 2018; 109: 513-22. ArticlePubMedPMCPDF
- 9. Heydt C, Wolwer CB, Velazquez Camacho O, et al. Detection of gene fusions using targeted next-generation sequencing: a comparative evaluation. BMC Med Genomics 2021; 14: 62.ArticlePubMedPMCPDF
- 10. Szurian K, Kashofer K, Liegl-Atzwanger B. Role of next-generation sequencing as a diagnostic tool for the evaluation of bone and soft-tissue tumors. Pathobiology 2017; 84: 323-38. ArticlePubMedPDF
- 11. Vendrell JA, Grand D, Rouquette I, et al. High-throughput detection of clinically targetable alterations using next-generation sequencing. Oncotarget 2017; 8: 40345-58. ArticlePubMedPMC
- 12. Bartels S, Persing S, Hasemeier B, Schipper E, Kreipe H, Lehmann U. Molecular analysis of circulating cell-free DNA from lung cancer patients in routine laboratory practice: a cross-platform comparison of three different molecular methods for mutation detection. J Mol Diagn 2017; 19: 722-32. ArticlePubMed
- 13. Underhill HR. Leveraging the fragment length of circulating tumour DNA to improve molecular profiling of solid tumour malignancies with next-generation sequencing: a pathway to advanced non-invasive diagnostics in precision oncology? Mol Diagn Ther 2021; 25: 389-408. ArticlePubMedPMCPDF
- 14. Song Z, Lu C, Xu CW, Zheng Z. Noncanonical gene fusions detected at the DNA level necessitate orthogonal diagnosis methods before targeted therapy. J Thorac Oncol 2021; 16: 344-8. ArticlePubMed
- 15. Li J, Lu H, Ng PK, et al. A functional genomic approach to actionable gene fusions for precision oncology. Sci Adv 2022; 8: eabm2382. ArticlePubMedPMC
- 16. Nikanjam M, Okamura R, Barkauskas DA, Kurzrock R. Targeting fusions for improved outcomes in oncology treatment. Cancer 2020; 126: 1315-21. ArticlePubMedPMCPDF
- 17. Polubothu S, McGuire N, Al-Olabi L, et al. Does the gene matter? Genotype-phenotype and genotype-outcome associations in congenital melanocytic naevi. Br J Dermatol 2020; 182: 434-43. ArticlePubMedPMCPDF
- 18. Dessars B, De Raeve LE, El Housni H, et al. Chromosomal translocations as a mechanism of BRAF activation in two cases of large congenital melanocytic nevi. J Invest Dermatol 2007; 127: 1468-70. ArticlePubMed
- 19. Mir A, Agim NG, Kane AA, Josephs SC, Park JY, Ludwig K. Giant congenital melanocytic nevus treated with trametinib. Pediatrics 2019; 143: e20182469. ArticlePubMedPDF
- 20. Molho-Pessach V, Hartshtark S, Merims S, et al. Giant congenital melanocytic naevus with a novel CUX1-BRAF fusion mutation treated with trametinib. Br J Dermatol 2022; 187: 1052-4. ArticlePubMedPMCPDF
- 21. Sampath AJ, Ruffolo AM, Miedema J, Googe PB, Thomas NE. Genetic abnormalities in congenital melanocytic nevi and their associated melanomas. JAAD Case Rep 2024; 45: 94-7. Article
- 22. Martin SB, Polubothu S, Bruzos AL, et al. Mosaic BRAF fusions are a recurrent cause of congenital melanocytic nevi targetable by MAPK pathway inhibition. J Invest Dermatol 2024; 144: 593-600. Article
- 23. Botton T, Yeh I, Nelson T, et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res 2013; 26: 845-51. ArticlePubMedPMC
- 24. Agrawal S, Guo R. Melanocytic neoplasm with novel EPS15:BRAF fusion and congenital features. Hum Pathol 2025; 165: 105790.ArticlePubMed
- 25. Roy SF, Agim NG, Mir A, et al. Congenital melanocytic naevi initiated by BRAF fusion oncogene with firmness, pruritus and desmoplastic stroma. Br J Dermatol 2025; 193: 232-9. ArticlePubMedPMCPDF
- 26. El-Rayes D, Wilson K, Maguiness S, Miller D, Cazzato G, Giubellino A. Congenital melanocytic nevus with neurocristic cutaneous hamartoma: a case report. Dermatopathology (Basel) 2025; 12: 12.ArticlePubMedPMC
- 27. Baltres A, Salhi A, Houlier A, et al. Malignant melanoma with areas of rhabdomyosarcomatous differentiation arising in a giant congenital nevus with RAF1 gene fusion. Pigment Cell Melanoma Res 2019; 32: 708-13. ArticlePubMedPDF
- 28. Martins da Silva V, Martinez-Barrios E, Tell-Marti G, et al. Genetic abnormalities in large to giant congenital nevi: beyond NRAS mutations. J Invest Dermatol 2019; 139: 900-8. ArticlePubMed
- 29. Vinyals A, Ferreres JR, Calbet-Llopart N, et al. Oncogenic properties via MAPK signaling of the SOX5-RAF1 fusion gene identified in a wild-type NRAS/BRAF giant congenital nevus. Pigment Cell Melanoma Res 2022; 35: 450-60. ArticlePubMedPDF
- 30. Houlier A, Pissaloux D, Tirode F, et al. RASGRF2 gene fusions identified in a variety of melanocytic lesions with distinct morphological features. Pigment Cell Melanoma Res 2021; 34: 1074-83. ArticlePubMedPDF
- 31. Korolkova OY, Widatalla SE, Whalen DS, et al. Reciprocal expression of annexin A6 and RasGRF2 discriminates rapidly growing from invasive triple negative breast cancer subsets. PLoS One 2020; 15: e0231711. ArticlePubMedPMC
- 32. Lu P, Chen J, Yan L, et al. RasGRF2 promotes migration and invasion of colorectal cancer cells by modulating expression of MMP9 through Src/Akt/NF-κB pathway. Cancer Biol Ther 2019; 20: 435-43. ArticlePubMedPMC
- 33. Nakagawa T, Kim Y, Kano J, et al. High expression of Ras-specific guanine nucleotide-releasing factor 2 (RasGRF2) in lung adenocarcinoma is associated with tumor invasion and poor prognosis. Pathol Int 2021; 71: 255-60. ArticlePubMedPMCPDF
- 34. Bahrami A, Lee S, Wu G, et al. Pigment-synthesizing melanocytic neoplasm with protein kinase C alpha (PRKCA) fusion. JAMA Dermatol 2016; 152: 318-22. ArticlePubMedPMC
- 35. Cohen JN, Joseph NM, North JP, Onodera C, Zembowicz A, LeBoit PE. Genomic analysis of pigmented epithelioid melanocytomas reveals recurrent alterations in PRKAR1A, and PRKCA genes. Am J Surg Pathol 2017; 41: 1333-46. Article
- 36. Perron E, Pissaloux D, Charon Barra C, et al. Melanocytic myxoid spindle cell tumor with ALK rearrangement (MMySTAR): report of 4 cases of a nevus variant with potential diagnostic challenge. Am J Surg Pathol 2018; 42: 595-603.
- 37. Moran JM, Le LP, Nardi V, et al. Identification of fusions with potential clinical significance in melanoma. Mod Pathol 2022; 35: 1837-47. ArticlePubMedPDF
- 38. Ibrahim E, Yang R, Nagarajan P, Sweeney K, Chen H, Roy-Chowdhuri S, et al. Clinicopathologic and molecular characterization of BRAF-fused melanomas: a retrospective analysis of 9 cases with 8 novel fusions detected by next-generation sequencing. Lab Invest 2025; 105(3 Suppl): 102669.
- 39. Turner JA, Bemis JGT, Bagby SM, et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene 2019; 38: 1296-308. ArticlePubMedPDF
- 40. Botton T, Talevich E, Mishra VK, et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep 2019; 29: 573-88. ArticlePubMedPMC
- 41. Hutchinson KE, Lipson D, Stephens PJ, et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 2013; 19: 6696-702. ArticlePubMedPMCPDF
- 42. Clark HE, Huang YY, Vance GH, Alomari AK. Fatal melanoma with a novel MYO5A-BRAF fusion and small associated conventional nevus: a case report and review of literature. J Cutan Pathol 2022; 49: 808-12. ArticlePubMedPMCPDF
- 43. Ross JS, Wang K, Chmielecki J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016; 138: 881-90. ArticlePubMed
- 44. Busam KJ, Shah KN, Gerami P, Sitzman T, Jungbluth AA, Kinsler V. Reduced H3K27me3 expression is common in nodular melanomas of childhood associated with congenital melanocytic nevi but not in proliferative nodules. Am J Surg Pathol 2017; 41: 396-404. ArticlePubMedPMC
- 45. Newell F, Wilmott JS, Johansson PA, et al. Whole-genome sequencing of acral melanoma reveals genomic complexity and diversity. Nat Commun 2020; 11: 5259.ArticlePubMedPMCPDF
- 46. Menzies AM, Yeh I, Botton T, Bastian BC, Scolyer RA, Long GV. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res 2015; 28: 607-10. ArticlePubMedPMCPDF
- 47. Kim HS, Jung M, Kang HN, et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017; 36: 3334-45. ArticlePubMedPDF
- 48. Perron E, Pissaloux D, Neub A, et al. Unclassified sclerosing malignant melanomas with AKAP9-BRAF gene fusion: a report of two cases and review of BRAF fusions in melanocytic tumors. Virchows Arch 2018; 472: 469-76. ArticlePubMedPDF
- 49. Gao Y, Yang RK, Curry JL, Torres-Cabala CA, Cho WC. Melanoma with RNF11::BRAF fusion: a novel fusion previously undescribed in melanoma. Am J Dermatopathol 2025; 47: 391-4. ArticlePubMed
- 50. Cho WC, Yang RK, Lenskaya V, Prieto VG. SBF1::BRAF-fused melanoma with prominent rhabdoid morphology and Reed-Sternberg-like cells: potential morphologic clues to BRAF-fused melanomas of non-Spitz lineage? Am J Dermatopathol 2025; 47: 733-5. ArticlePubMed
- 51. Johnson DB, Nebhan CA, Noel MS. MEK inhibitors in non-V600 BRAF mutations and fusions. Oncotarget 2020; 11: 3900-3. ArticlePubMedPMC
- 52. Williams EA, Shah N, Montesion M, et al. Melanomas with activating RAF1 fusions: clinical, histopathologic, and molecular profiles. Mod Pathol 2020; 33: 1466-74. ArticlePubMedPMCPDF
- 53. Khaddour K, Haq R, Buchbinder EI, et al. Targeting RAF1 gene fusions with MEK inhibition in metastatic melanoma. Oncologist 2025; 30: oyae297.ArticlePubMedPMCPDF
- 54. Kim KB, Semrad T, Schrock AB, et al. Significant clinical response to a MEK inhibitor therapy in a patient with metastatic melanoma harboring an RAF1 fusion. JCO Precis Oncol 2018; 2: 1-6. ArticlePubMedPMC
- 55. McEvoy CR, Xu H, Smith K, et al. Profound MEK inhibitor response in a cutaneous melanoma harboring a GOLGA4-RAF1 fusion. J Clin Invest 2019; 129: 1940-5. Article
- 56. Boileau M, Descarpentries C, Delzenne G, et al. Clinical response under MEK inhibitor alone in metastatic melanoma with a novel fusion involving the RAF1 gene. Melanoma Res 2023; 33: 247-51. Article
- 57. Hayward NK, Wilmott JS, Waddell N, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017; 545: 175-80. ArticlePubMed
- 58. Pacaud A, Amintas S, Boussemart L, Cappellen D, Gerard E. A case of multi-metastatic melanoma with RAF1 fusion: a surprising response to anti-MEK therapy. Eur J Cancer 2021; 147: 161-3. ArticlePubMed
- 59. Do J, Yang RK, Curry JL, Cho WC. Fatal melanoma with MAP4::RAF1 fusion: expanding the clinicopathologic and prognostic spectrum of RAF1-fused melanomas. Am J Dermatopathol 2026; 48: 135-9. ArticlePubMed
- 60. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun 2014; 5: 4846.ArticlePDF
- 61. Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010; 16: 793-8. ArticlePubMedPMC
- 62. Jain P, Fierst TM, Han HJ, et al. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 2017; 36: 6348-58. ArticlePubMedPMCPDF
- 63. Lezcano C, Shoushtari AN, Ariyan C, Hollmann TJ, Busam KJ. Primary and metastatic melanoma with NTRK fusions. Am J Surg Pathol 2018; 42: 1052-8. ArticlePubMedPMC
- 64. Couts KL, Bemis J, Turner JA, et al. ALK inhibitor response in melanomas expressing EML4-ALK fusions and alternate ALK isoforms. Mol Cancer Ther 2018; 17: 222-31. ArticlePubMedPMCPDF
- 65. Perkins IU, Tan SY, McCalmont TH, et al. Melanoma in infants, caused by a gene fusion involving the anaplastic lymphoma kinase (ALK). Pigment Cell Melanoma Res 2024; 37: 6-14. ArticlePubMedPMC
- 66. Shaker N, Phelps R, Niedt G, et al. BRCA1-associated protein-1 inactivated melanoma arising in a pre-existing nevus with ALK fusion and low tumor mutational burden. Am J Dermatopathol 2025; 47: 264-8. ArticlePubMed
- 67. Lee J, Lee J, Hong SD, Jang KT, Lee SJ. FGFR3-TACC3: a novel gene fusion in malignant melanoma. Precis Future Med 2018; 2: 71-5. ArticlePDF
- 68. Lehmann BD, Shaver TM, Johnson DB, et al. Identification of targetable recurrent MAP3K8 rearrangements in melanomas lacking known driver mutations. Mol Cancer Res 2019; 17: 1842-53. ArticlePubMedPMCPDF
- 69. Liang WS, Hendricks W, Kiefer J, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res 2017; 27: 524-32. ArticlePubMedPMC
- 70. Wang J, Algarin YA, El-Sayed I, Bastian B, Yeh I. Mucosal melanoma with MAP3K8 gene rearrangement. In: The American Society of Dermatopathology, 61st Annual Meeting; 2024 Nov 4-10; Chicago, IL, USA.
- 71. Wang M, Banik I, Shain AH, Yeh I, Bastian BC. Integrated genomic analyses of acral and mucosal melanomas nominate novel driver genes. Genome Med 2022; 14: 65.ArticlePubMedPMCPDF
- 72. Williams EA, Montesion M, Shah N, et al. Melanoma with in-frame deletion of MAP2K1: a distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod Pathol 2020; 33: 2397-406. ArticlePubMedPMC
- 73. Ebbelaar CF, Jansen AM, Speet LC, et al. Clinical outcomes and genomic profiles of MAP2K1-mutated primary cutaneous melanocytic tumours. EBioMedicine 2025; 114: 105643.ArticlePubMedPMC
- 74. Furney SJ, Turajlic S, Stamp G, et al. The mutational burden of acral melanoma revealed by whole-genome sequencing and comparative analysis. Pigment Cell Melanoma Res 2014; 27: 835-8. ArticlePubMed
- 75. Ibrahim E, Yang RK, Curry JL, Cho WC. BCR::ZNF711 and BCR::CYLC2 fusions: novel BCR fusions expanding the molecular spectrum of gene fusions in melanoma. Am J Dermatopathol 2024; 46: 797-9. ArticlePubMed
- 76. Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA. The 2018 World Health Organization classification of cutaneous, mucosal, and uveal melanoma: detailed analysis of 9 distinct subtypes defined by their evolutionary pathway. Arch Pathol Lab Med 2020; 144: 500-22. ArticlePubMedPDF
- 77. Hagstrom M, Fumero-Velazquez M, Dhillon S, Olivares S, Gerami P. An update on genomic aberrations in Spitz naevi and tumours. Pathology 2023; 55: 196-205. ArticlePubMed
- 78. Delsupehe L, Steelandt T, Lemahieu J, et al. Novel gene fusion discovery in Spitz tumours and its relevance in diagnostics. Virchows Arch 2024; 485: 269-79. ArticlePubMedPDF
- 79. Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014; 5: 3116.ArticlePubMedPMCPDF
- 80. de la Fouchardiere A, Mazzei ME, Pastor M, Forster AM, Prieto VG. Spitz tumours and mimickers. Virchows Arch 2025; 486: 143-64. ArticlePubMedPDF
- 81. Sunshine JC, Kim D, Zhang B, et al. Melanocytic neoplasms with MAP2K1 in frame deletions and Spitz morphology. Am J Dermatopathol 2020; 42: 923-31. ArticlePubMedPMC
- 82. Goto K, Pissaloux D, Fraitag S, et al. RASGRF1-rearranged cutaneous melanocytic neoplasms with spitzoid cytomorphology: a clinicopathologic and genetic study of 3 cases. Am J Surg Pathol 2022; 46: 655-63. ArticlePubMed
- 83. Urso C. Melanocytic skin neoplasms: what lesson from genomic aberrations? Am J Dermatopathol 2019; 41: 623-9. ArticlePubMed
- 84. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science 2013; 339: 957-9. ArticlePubMedPMC
- 85. Quan VL, Zhang B, Zhang Y, et al. Integrating next-generation sequencing with morphology improves prognostic and biologic classification of Spitz neoplasms. J Invest Dermatol 2020; 140: 1599-608. ArticlePubMed
- 86. Barr RJ, Morales RV, Graham JH. Desmoplastic nevus: a distinct histologic variant of mixed spindle cell and epithelioid cell nevus. Cancer 1980; 46: 557-64. ArticlePubMed
- 87. Lezcano CM, Yeh I, Eslamdoost N, et al. Expanding the spectrum of microscopic and cytogenetic findings associated with Spitz tumors with 11p gains. Am J Surg Pathol 2021; 45: 277-85. ArticlePubMedPMC
- 88. Yeh I, Botton T, Talevich E, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 2015; 6: 7174.ArticlePubMedPMCPDF
- 89. Amin SM, Haugh AM, Lee CY, et al. A comparison of morphologic and molecular features of BRAF, ALK, and NTRK1 fusion spitzoid neoplasms. Am J Surg Pathol 2017; 41: 491-8. ArticlePubMed
- 90. Busam KJ, Kutzner H, Cerroni L, Wiesner T. Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am J Surg Pathol 2014; 38: 925-33. ArticlePubMedPMC
- 91. Salah HT, Yang RK, Roy-Chowdhuri S, et al. Spitz melanocytic neoplasms with MLPH::ALK fusions: report of two cases with previously unreported features and literature review. J Cutan Pathol 2024; 51: 407-14. ArticlePubMed
- 92. Kastnerova L, Martinek P, Grossmann P, et al. A clinicopathological study of 29 spitzoid melanocytic lesions with ALK fusions, including novel fusion variants, accompanied by fluorescence in situ hybridization analysis for chromosomal copy number changes, and both TERT promoter and next-generation sequencing mutation analysis. Am J Dermatopathol 2020; 42: 578-92. ArticlePubMed
- 93. Bahrani E, Kunder CA, Teng JM, et al. Spitz nevus with EHBP1-ALK fusion and distinctive membranous localization of ALK. J Cutan Pathol 2022; 49: 584-8. ArticlePubMedPDF
- 94. Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015; 39: 581-91. Article
- 95. Chung CT, Marrano P, Swanson D, Dickson BC, Thorner PS. Fusion of ALK to the melanophilin gene MLPH in pediatric Spitz nevi. Hum Pathol 2019; 87: 57-64. ArticlePubMed
- 96. Fujimoto M, Togashi Y, Matsuzaki I, et al. A case report of atypical Spitz tumor harboring a novel MLPH-ALK gene fusion with discordant ALK immunohistochemistry results. Hum Pathol 2018; 80: 99-103. ArticlePubMed
- 97. Schoelinck J, Pissaloux D, Mouthon M, Vergara R, de la Fouchardiere A. Clinical, histological and genetic correlations in melanocytic tumours with chromosomal rearrangements. Ann Pathol 2025; 45: 3-14. ArticlePubMed
- 98. Raghavan SS, Peternel S, Mully TW, et al. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod Pathol 2020; 33: 1122-34. ArticlePubMedPMCPDF
- 99. Cho WC, Prieto VG, Yang RK. Spitz melanoma with SLC20A1::ALK fusion: a novel fusion previously undescribed in Spitz melanocytic neoplasm. Am J Dermatopathol 2024; 46: 700-3. ArticlePubMed
- 100. Frederico IK, Mesbah Ardakani N, Ryan AL, Cowley MJ, Wood BA. Spitz melanoma of childhood with a novel promoter hijacking anaplastic lymphoma kinase (C2orf42-ALK) rearrangement. Am J Dermatopathol 2021; 43: 972-5. ArticlePubMed
- 101. Pappo AS, McPherson V, Pan H, et al. A prospective, comprehensive registry that integrates the molecular analysis of pediatric and adolescent melanocytic lesions. Cancer 2021; 127: 3825-31. ArticlePubMedPMCPDF
- 102. Gerami P, Kim D, Compres EV, et al. Clinical, morphologic, and genomic findings in ROS1 fusion Spitz neoplasms. Mod Pathol 2021; 34: 348-57. ArticlePubMedPMCPDF
- 103. Donati M, Kastnerova L, Martinek P, et al. Spitz tumors with ROS1 fusions: a clinicopathological study of 6 cases, including FISH for chromosomal copy number alterations and mutation analysis using next-generation sequencing. Am J Dermatopathol 2020; 42: 92-102. ArticlePubMed
- 104. Lee S, Barnhill RL, Dummer R, et al. TERT promoter mutations are predictive of aggressive clinical behavior in patients with spitzoid melanocytic neoplasms. Sci Rep 2015; 5: 11200.ArticlePubMedPMCPDF
- 105. Robertson SJ, Orme L, Teixeira R, et al. Evaluation of crizotinib treatment in a patient with unresectable GOPC-ROS1 fusion agminated Spitz nevi. JAMA Dermatol 2021; 157: 836-41. ArticlePubMedPMC
- 106. Church AJ, Moustafa D, Pinches RS, Hawryluk EB, Schmidt BA. Genomic comparison of malignant melanoma and atypical Spitz tumor in the pediatric population. Pediatr Dermatol 2022; 39: 409-19. ArticlePDF
- 107. Raghavan SS, Kapler ES, Dinges MM, Bastian BC, Yeh I. Eruptive Spitz nevus, a striking example of benign metastasis. Sci Rep 2020; 10: 16216.ArticlePDF
- 108. Cesinaro AM, Gallo G, Manfredini S, Maiorana A, Bettelli SR. ROS1 pattern of immunostaining in 11 cases of spitzoid tumour: comparison with histopathological, fluorescence in-situ hybridisation and next-generation sequencing analysis. Histopathology 2021; 79: 966-74. ArticlePubMedPDF
- 109. Couts KL, McCoach CE, Murphy D, et al. Acral lentiginous melanoma harboring a ROS1 gene fusion with clinical response to entrectinib. JCO Precis Oncol 2017; 1: 1-7. ArticlePubMed
- 110. Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol 2019; 30: viii5-15. ArticlePubMedPMCPDF
- 111. Yeh I, Busam KJ, McCalmont TH, et al. Filigree-like rete ridges, lobulated nests, rosette-like structures, and exaggerated maturation characterize Spitz tumors with NTRK1 fusion. Am J Surg Pathol 2019; 43: 737-46. ArticlePubMed
- 112. Khotskaya YB, Holla VR, Farago AF, Mills Shaw KR, Meric-Bernstam F, Hong DS. Targeting TRK family proteins in cancer. Pharmacol Ther 2017; 173: 58-66. ArticlePubMed
- 113. Cappellesso R, Nozzoli F, Zito Marino F, et al. NTRK gene fusion detection in atypical Spitz tumors. Int J Mol Sci 2021; 22: 12332.ArticlePubMedPMC
- 114. Yin L, Shi C, He X, et al. NTRK-rearranged spindle cell neoplasms: a clinicopathological and molecular study of 13 cases with peculiar characteristics at one of the largest institutions in China. Pathology 2023; 55: 362-74. ArticlePubMed
- 115. Dal Pozzo CA, Cappellesso R. The morpho-molecular landscape of Spitz neoplasms. Int J Mol Sci 2022; 23: 4211.ArticlePubMedPMC
- 116. Kervarrec T, Pissaloux D, Tirode F, et al. Morphologic features in a series of 352 Spitz melanocytic proliferations help predict their oncogenic drivers. Virchows Arch 2022; 480: 369-82. ArticlePubMedPDF
- 117. Kiuru M, Jungbluth A, Kutzner H, Wiesner T, Busam KJ. Spitz Tumors: Comparison of histological features in relationship to immunohistochemical staining for ALK and NTRK1. Int J Surg Pathol 2016; 24: 200-6. ArticlePubMedPDF
- 118. Sekoranja D, Pizem J, Luzar B. An update on molecular genetic aberrations in Spitz melanocytic proliferations: correlation with morphological features and biological behavior. Acta Med Acad 2021; 50: 157-74. ArticlePubMedPDF
- 119. Forschner A, Forchhammer S, Bonzheim I. NTRK gene fusions in melanoma: detection, prevalence and potential therapeutic implications. J Dtsch Dermatol Ges 2020; 18: 1387-92. ArticlePubMedPDF
- 120. Cazzato G, Colagrande A, Resta L, et al. LMNA::NTRK1 and PRDX1::NTRK1 atypical Spitz tumor: a report of two additional cases with histological, immunohistochemical, and molecular insights. Am J Dermatopathol 2025; 47: 22-4. ArticlePubMed
- 121. Yeh I, Tee MK, Botton T, et al. NTRK3 kinase fusions in Spitz tumours. J Pathol 2016; 240: 282-90. ArticlePubMedPMC
- 122. Lee CY, Sholl LM, Zhang B, et al. Atypical spitzoid neoplasms in childhood: a molecular and outcome study. Am J Dermatopathol 2017; 39: 181-6. ArticlePubMed
- 123. Goto K, Pissaloux D, Tirode F, de la Fouchardiere A. Spitz nevus with a novel TFG-NTRK2 fusion: the first case report of NTRK2-rearranged Spitz/Reed nevus. J Cutan Pathol 2021; 48: 1193-6. ArticlePubMedPDF
- 124. Mansour B, Vanecek T, Kastnerova L, Nosek D, Kazakov DV, Donati M. Spitz tumor with SQSTM1::NTRK2 fusion: a clinicopathological study of 5 cases. Am J Dermatopathol 2023; 45: 306-10. ArticlePubMed
- 125. VandenBoom T, Quan VL, Zhang B, et al. Genomic fusions in pigmented spindle cell nevus of reed. Am J Surg Pathol 2018; 42: 1042-51. ArticlePubMed
- 126. Wang L, Busam KJ, Benayed R, et al. Identification of NTRK3 fusions in childhood melanocytic neoplasms. J Mol Diagn 2017; 19: 387-96. ArticlePubMedPMC
- 127. de la Fouchardiere A, Tee MK, Peternel S, et al. Fusion partners of NTRK3 affect subcellular localization of the fusion kinase and cytomorphology of melanocytes. Mod Pathol 2021; 34: 735-47. ArticlePubMedPMCPDF
- 128. Della Mura M, Sorino J, Colagrande A, et al. Atypical Spitz tumor/Spitz melanocytoma revealing a VIM::NTRK3 gene fusion: clinicopathological correlation of a unique molecular finding. Am J Dermatopathol 2025; 47: 886-90. ArticlePubMed
- 129. Donati M, Goutas D, Pissaloux D, et al. Clinical, morphologic, and genomic findings in Spitz tumors with RET fusion: a series of 31 cases. Mod Pathol 2025; 38: 100740.ArticlePubMed
- 130. Kim D, Compres EV, Zhang B, et al. A series of RET fusion Spitz neoplasms with plaque-like silhouette and dyscohesive nesting of epithelioid melanocytes. Am J Dermatopathol 2021; 43: 243-51. ArticlePubMed
- 131. Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther 2011; 10: 2298-308. ArticlePDF
- 132. Zarabi SK, Azzato EM, Tu ZJ, et al. Targeted next generation sequencing (NGS) to classify melanocytic neoplasms. J Cutan Pathol 2020; 47: 691-704. ArticlePubMedPDF
- 133. Feldman T, Zaaroura H, Abaya HH, Zohar Y, Bergman R. Congenital Spitz melanocytoma with activating ZKSCAN1::MET kinase fusion. J Cutan Pathol 2025; 52: 617-21. ArticlePubMed
- 134. Roy S, Milante R, Busam K, de la Fouchardiere A, Yeh I. MET-fused melanocytic Spitz tumors: 18 cases with identifiable histomorphological and genetic features. Lab Invest 2025; 105(3 Suppl): 102693.
- 135. Wu G, Barnhill RL, Lee S, et al. The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod Pathol 2016; 29: 359-69. ArticlePubMedPMCPDF
- 136. Kim D, Khan AU, Compres EV, et al. BRAF fusion Spitz neoplasms: clinical morphological, and genomic findings in six cases. J Cutan Pathol 2020; 47: 1132-42. ArticlePubMedPDF
- 137. Donati M, Kastnerova L, Ptakova N, Michal M, Kazakov DV. Polypoid atypical Spitz tumor with a fibrosclerotic stroma, CLIP2-BRAF fusion, and homozygous loss of 9p21. Am J Dermatopathol 2020; 42: 204-7. ArticlePubMed
- 138. Newman S, Fan L, Pribnow A, et al. Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat Med 2019; 25: 597-602. PMC
- 139. Roy SF, Bastian BC, Maguiness S, et al. Multiple desmoplastic Spitz nevi with BRAF fusions in a patient with ring chromosome 7 syndrome. Pigment Cell Melanoma Res 2021; 34: 987-93. ArticlePubMedPMCPDF
- 140. Sorino J, Della Mura M, Ingravallo G, Colagrande A, Cazzato G. TAX1BP1::BRAF fusion in BRAF-mutated atypical Spitz tumor/Spitz melanocytoma: a novel molecular finding with histopathological insights. Am J Dermatopathol 2025; 47: 568-9. ArticlePubMed
- 141. Sharma N, Patel P, Chen A, et al. The clinical, morphologic, and molecular spectrum of BRAF fusion Spitz tumors. Am J Surg Pathol 2024; 48: 1588-99. ArticlePubMed
- 142. Roy SF, Milante R, Pissaloux D, et al. Spectrum of melanocytic tumors harboring BRAF gene fusions: 58 cases with histomorphologic and genetic correlations. Mod Pathol 2023; 36: 100149.ArticlePubMed
- 143. Hiraki T, Hirakawa S, Otsuki Y, Kajimoto K, Goto K, Serizawa M. Fatal Spitz melanoma with MAD1L1::BRAF fusion: a case report and literature review. J Cutan Pathol 2025; 52: 199-205. ArticlePubMed
- 144. Donati M, Nosek D, Olivares S, et al. Spitz tumor with RAF1 fusion: a report of 3 cases. Ann Diagn Pathol 2023; 67: 152215.ArticlePubMed
- 145. Houlier A, Pissaloux D, Masse I, et al. Melanocytic tumors with MAP3K8 fusions: report of 33 cases with morphological-genetic correlations. Mod Pathol 2020; 33: 846-57. ArticlePubMedPDF
- 146. Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468: 968-72. ArticlePubMedPMC
- 147. Newman S, Pappo A, Raimondi S, Zhang J, Barnhill R, Bahrami A. Pathologic characteristics of Spitz melanoma with MAP3K8 fusion or truncation in a pediatric cohort. Am J Surg Pathol 2019; 43: 1631-7. ArticlePubMed
- 148. Quan VL, Zhang B, Mohan LS, et al. Activating structural alterations in MAPK genes are distinct genetic drivers in a unique subgroup of Spitzoid neoplasms. Am J Surg Pathol 2019; 43: 538-48. Article
- 149. Sibira R, Vu A, Giubellino A, Murugan P. Spitz melanoma with MAP3K8::ABLIM1 rearrangement: a case report with review of the literature. Diagn Pathol 2024; 19: 133.ArticlePubMedPMCPDF
- 150. Donati M, Nosek D, Waldenback P, et al. MAP2K1-mutated melanocytic neoplasms with a SPARK-like morphology. Am J Dermatopathol 2021; 43: 412-7. ArticlePubMed
- 151. Kerckhoffs KG, Aallali T, Ambarus CA, Sigurdsson V, Jansen AM, Blokx WA. Expanding spectrum of "spitzoid" lesions: a small series of 4 cases with MAP2K1 mutations. Virchows Arch 2021; 479: 195-202. ArticlePubMedPMCPDF
- 152. Kervarrec T, Pissaloux D, Chokri I, Tirode F, de la Fouchardiere A. MST1R/RON fusion as a potential oncogenic driver in Spitz tumours. Pathology 2024; 56: 1051-3. ArticlePubMed
- 153. Goto K, Pissaloux D, Durand L, Tirode F, Guillot B, de la Fouchardiere A. Novel three-way complex rearrangement of TRPM1-PUM1-LCK in a case of agminated Spitz nevi arising in a giant congenital hyperpigmented macule. Pigment Cell Melanoma Res 2020; 33: 767-72. ArticlePubMedPDF
- 154. Giubellino A, He Y, Munro SA, et al. Gene expression profile of benign, intermediate, and malignant Spitz and spitzoid melanocytic lesions. Cancers (Basel) 2024; 16: 1798.ArticlePubMedPMC
- 155. Scarfo F, Brunetto E, Magliacane G, Pecciarini L, Ferrara G, Rizzo N. Spitz spindle cell/reed nevus with SQSTM1::NTRK2 fusion and atypical features in an older male patient: a case report and review of literature. J Cutan Pathol 2025; 52: 367-73. ArticlePubMedPDF
- 156. Mohan LS, Khan AU, Zhang B, et al. Retrospective cohort: genomic differences between pigmented spindle cell nevi of reed and reed-like melanomas. Am J Dermatopathol 2020; 42: 641-7. ArticlePubMed
- 157. Zembowicz A, Carney JA, Mihm MC. Pigmented epithelioid melanocytoma: a low-grade melanocytic tumor with metastatic potential indistinguishable from animal-type melanoma and epithelioid blue nevus. Am J Surg Pathol 2004; 28: 31-40. ArticlePubMed
- 158. Zembowicz A, Knoepp SM, Bei T, et al. Loss of expression of protein kinase a regulatory subunit 1alpha in pigmented epithelioid melanocytoma but not in melanoma or other melanocytic lesions. Am J Surg Pathol 2007; 31: 1764-75. ArticlePubMed
- 159. Isales MC, Mohan LS, Quan VL, et al. Distinct genomic patterns in pigmented epithelioid melanocytoma: a molecular and histologic analysis of 16 cases. Am J Surg Pathol 2019; 43: 480-8. ArticlePubMed
- 160. Zaaroura H, Cyrenne B, Somers GR, et al. Two congenital cases of pigmented epithelioid melanocytoma with unique clinical and genetic features. Dermatol Online J 2023; 29: 3.ArticlePubMed
- 161. Friedman BJ, Hernandez S, Fidai C, et al. A pediatric case of pigmented epithelioid melanocytoma with chromosomal copy number alterations in 15q and 17q and a novel NTRK3-SCAPER gene fusion. J Cutan Pathol 2020; 47: 70-5. ArticlePubMedPDF
- 162. Scollan ME, Yamashiro DJ, Niedt GW, Garzon MC. Novel CD63-PRKCB fusion in a case of pigmented epithelioid melanocytoma. Pediatr Dermatol 2022; 39: 322-3. ArticlePubMedPDF
- 163. Donati M, Kastnerova L, Cempirkova D, Vanecek T, Michal M, Kazakov DV. Vulvar pigmented epithelioid melanocytoma with a novel HTT-PKN1 fusion: a case report. Am J Dermatopathol 2020; 42: 544-6. ArticlePubMed
- 164. Li A, Umphress B, Dehner C, et al. Genomic and transcriptomic characterization of protein kinase C fusion melanocytic neoplasms with distinctive hypopigmented histomorphology: a single-institution study. J Cutan Pathol 2025; 52: 432-41. ArticlePubMedPMC
- 165. de la Fouchardiere A, Pissaloux D, Houlier A, et al. Histologic and genetic features of 51 melanocytic neoplasms with protein kinase C fusion genes. Mod Pathol 2023; 36: 100286.ArticlePubMed
- 166. Zhao J, Lampley N, Benton S, et al. Next-generation sequencing reveals a new class of melanocytic neoplasms with hybrid genomic features of PEM including protein kinase R 1 alpha gene inactivation and Spitz tumor-defining protein kinase fusions. Am J Dermatopathol 2022; 44: 568-74. ArticlePubMed
- 167. Kervarrec T, Lo Bello G, Pissaloux D, et al. GRM1 gene fusions as an alternative molecular driver in blue nevi and related melanomas. Mod Pathol 2023; 36: 100264.ArticlePubMed
- 168. Yeh I, Lang UE, Durieux E, et al. Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat Commun 2017; 8: 644.ArticlePubMedPMCPDF
- 169. Parra O, Linos K. Cutaneous melanocytic tumor with CRTC1::TRIM11 fusion: review of the literature of a potentially novel entity. Biology (Basel) 2021; 10: 1286.ArticlePubMedPMC
- 170. Cellier L, Perron E, Pissaloux D, et al. Cutaneous melanocytoma with CRTC1-TRIM11 fusion: report of 5 cases resembling clear cell sarcoma. Am J Surg Pathol 2018; 42: 382-91. ArticlePubMed
- 171. Miyagi Y, Senzaki H, Kato I, Kakuda Y, Goto K. A case of a CRTC1::TRIM11 cutaneous tumor with venous and lymphatic invasion and lymph node metastasis. Cureus 2025; 17: e83821.ArticlePubMedPMC
- 172. Tseng C, Hein EC, Smith SM, King I, Saibil S, Saeed Kamil Z. Ulcerated CRTC1::TRIM11 cutaneous tumor with metastases. J Cutan Pathol 2024; 51: 735-41. ArticlePubMed
- 173. Bontoux C, Baroudjian B, Le Maignan C, et al. CRTC1-TRIM11 fusion in a case of metastatic clear cell sarcoma: are CRTC1-TRIM11 fusion-bearing tumors melanocytomas or clear cell sarcomas? Am J Surg Pathol 2019; 43: 861-3. ArticlePubMed
- 174. Kashima J, Motoi T, Nishimaki M, et al. A case report of cutaneous melanocytoma with CRTC1-TRIM11 fusion: Is CMCT distinct from clear cell sarcoma of soft tissue? Pathol Int 2019; 69: 496-501. ArticlePubMedPDF
- 175. Ko JS, Wang L, Billings SD, et al. CRTC1-TRIM11 fusion defined melanocytic tumors: a series of four cases. J Cutan Pathol 2019; 46: 810-8. ArticlePubMedPDF
- 176. Ko JS, Lemahieu J, Billings SD, et al. MED15::ATF1-rearranged tumor: a novel cutaneous tumor with melanocytic differentiation. Mod Pathol 2024; 37: 100438.ArticlePubMed
- 177. de la Fouchardiere A, Pissaloux D, Tirode F, Karanian M, Fletcher CD, Hanna J. Clear cell tumor with melanocytic differentiation and ACTIN-MITF translocation: report of 7 cases of a novel entity. Am J Surg Pathol 2021; 45: 962-8. ArticlePubMed
- 178. Kalmykova AV, Baranovska-Andrigo V, Michal M. Update on cutaneous mesenchymal tumors in the 5th edition of WHO classification of skin tumors with an emphasis on new fusion-associated neoplasms. Virchows Arch 2024; 485: 777-92. ArticlePubMedPMCPDF
- 179. Kalmykova A, Mosaieby E, Kacerovska D, et al. MITF::CREM-rearranged tumor: a novel group of cutaneous tumors with melanocytic differentiation. Virchows Arch 2023; 483: 569-75. ArticlePubMedPDF
- 180. Bigot NJ, Neyaz A, Naous R, et al. Rapidly enlarging ACTIN::MITF rearranged clear cell tumour with melanocytic differentiation. Histopathology 2025; 86: 829-32. Article
- 181. Alexandrescu S, Imamovic-Tuco A, Janeway K, Hanna J. Clear cell tumor with melanocytic differentiation and MITF::CREM translocation. J Cutan Pathol 2023; 50: 619-22. ArticlePubMed
- 182. de la Fouchardiere A, Pissaloux D, Tirode F, Hanna J. Clear cell tumor with melanocytic differentiation and MITF-CREM translocation: a novel entity similar to clear cell sarcoma. Virchows Arch 2021; 479: 841-6. ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-