 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 53(2); 2019 > Article
-
Original Article
Guanabenz Acetate Induces Endoplasmic Reticulum Stress–Related Cell Death in Hepatocellular Carcinoma Cells -
Hyo Jeong Kang1,*
 , Hyang Sook Seol2,*, Sang Eun Lee2, Young-Ah Suh2, Jihun Kim1, Se Jin Jang1,2, Eunsil Yu1,3
, Hyang Sook Seol2,*, Sang Eun Lee2, Young-Ah Suh2, Jihun Kim1, Se Jin Jang1,2, Eunsil Yu1,3 -
Journal of Pathology and Translational Medicine 2019;53(2):94-103.
DOI: https://doi.org/10.4132/jptm.2019.01.14
Published online: January 16, 2019
1Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
2Asan institute for Life Science, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
3Asan Liver Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
-
Corresponding Author Se Jin Jang, MD, PhD Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-5608 Fax: +82-2-472-7898 E-mail: jangsejin@amc.seoul.kr
Eunsil Yu, MD, PhD Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-4552 Fax: +82-2-472-7898 E-mail: d890075@gmail.com *Hyo Jeong Kang and Hyang Sook Seol contributed equally to this work.
© 2019 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Development of chemotherapeutics for the treatment of advanced hepatocellular carcinoma (HCC) has been lagging. Screening of candidate therapeutic agents by using patient-derived preclinical models may facilitate drug discovery for HCC patients.
-
Methods
- Four primary cultured HCC cells from surgically resected tumor tissues and six HCC cell lines were used for high-throughput screening of 252 drugs from the Prestwick Chemical Library. The efficacy and mechanisms of action of the candidate anti-cancer drug were analyzed via cell viability, cell cycle assays, and western blotting.
-
Results
- Guanabenz acetate, which has been used as an antihypertensive drug, was screened as a candidate anti-cancer agent for HCC through a drug sensitivity assay by using the primary cultured HCC cells and HCC cell lines. Guanabenz acetate reduced HCC cell viability through apoptosis and autophagy. This occurred via inhibition of growth arrest and DNA damage-inducible protein 34, increased phosphorylation of eukaryotic initiation factor 2α, increased activating transcription factor 4, and cell cycle arrest.
-
Conclusions
- Guanabenz acetate induces endoplasmic reticulum stress–related cell death in HCC and may be repositioned as an anti-cancer therapeutic agent for HCC patients.
- Ethical approval
- This study was approved by the Institutional Review Board of Asan Medical Center with a waiver of informed consent (IRB No. 2012-0112).
- Patients and samples
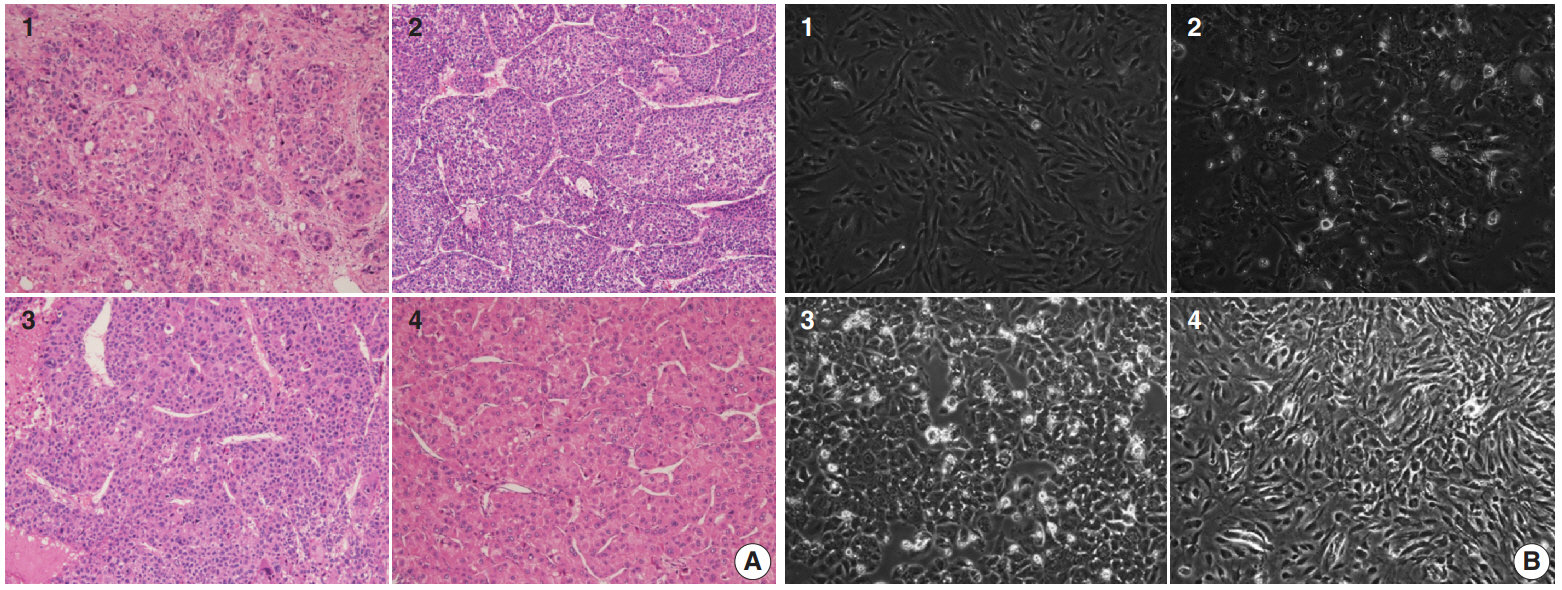
- Four patients with HCC, who were confirmed to have the disease via liver protocol dynamic computed tomography scans at Asan Medical Center, Seoul, Korea, were selected to establish primary cultured cells. A portion of the tumor tissues obtained from surgical resection were fixed in cold 2% formaldehyde for 4 hours and embedded in paraffin at 56°C. Sections from the paraffin blocks (4-μm thick) were stained with hematoxylin and eosin (H&E). All H&E slides were reviewed by two pathologists (E.Y. and H.J.K.) who were blinded to the clinical information.
- Primary culture of HCC cells
- The fresh tissues of the four cases obtained during surgery were put into a tube containing Dulbecco’s modified Eagle medium: nutrient mixture F-12 (DMEM/F12; Sigma-Aldrich, St. Louis, MO, USA), with penicillin and streptomycin, and were transported to the tissue culture room. After removing normal liver tissue and connective tissue, tumor tissues were minced with scissors and subsequently digested with 0.1% type IV collagenase (Sigma-Aldrich) in a shaking incubator for 60 minutes at 37°C. After incubation, tissues were washed three to four times with DMEM/F12 containing 10% fetal bovine serum (FBS; Sigma-Aldrich). The HCC pellets obtained after centrifugation were plated on collagen type I dishes and incubated at 37°C in a 5% CO2 atmosphere. To favor the adhesion and growth of epithelial tumor cells, hepatocyte basal media containing human epidermal growth factor, transferrin, hydrocortisone, bovine serum albumin, ascorbic acid, GA-1000 (gentamicin and amphotericin B), insulin, 10% FBS, and a Triiodothyronine-SingleQuots kit (Biocompare, South San Francisco, CA, USA) were used to culture primary HCC tumor cells. The cultured HCC cells were harvested and stored in liquid nitrogen.
- MTT cell proliferation assay/drug sensitivity assay
- Two hundred and fifty-two small molecules from the Prestwick Chemical Library (Prestwick Chemical, Illkirch, France) were blindly selected for screening of anti-cancer effect. The primary cultured HCC cells (103 cells/well) were seeded in collagen type I coated 96-well plates. Following a 24-hour incubation at 37°C, the cells were treated with 252 small molecules (20 μM each). After 72 hours, 50 mg/mL of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reagent were added to each well, and the plates were incubated for 4 hours at 37°C. After the reduced formazan precipitate was dissolved in dimethyl sulfoxide, the absorbance at 540 nm was measured using a microplate reader (EL800, BioTek, Winooski, VT, USA). The concentration of candidate molecules to inhibit 50% of cell viability (IC50 value) was calculated using CalcuSyn Software (Biosoft, Cambridge, UK). These results were validated in six stable HCC cell lines (i.e., SNU398, SNU423, SNU449, SNU475, Hep3B, and Huh7). Five HCC cell lines (i.e., SNU398, SNU423, SNU 449, SNU475, and Huh7) and one HCC cell line (Hep3B) were respectively grown in RPMI 1640 medium (Sigma-Aldrich) and Minimum Essential Medium (Sigma-Aldrich), both supplemented with 10% FBS, penicillin, and streptomycin.
- Small interfering RNA transfection
- Hep3B and Huh7 HCC cells were treated with small interfering RNA (siRNA) specific to GADD34 and non-specific siRNA (Genolution Pharmaceuticals Inc., Seoul, Korea). Targeting sequences of siRNA were as follows: siGADD34-1, 5'-GGAUAAGGAAGAUGAUUCAUU-3'; siGADD34-2, 5'-GCCUAUAAUUUAUUAACUAUU-3'; and as a negative control, non-specific siRNA, 5'-CCUCGUGCCGUUCCAUCAGGUAGUU-3'. HCC cells were transiently transfected with GADD34 siRNA or non-specific siRNA with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Six hours later, the medium was replaced with regular culture medium, and the cells were incubated. The efficiency of silencing was assessed via western blotting 48 hours after transfection.
- Western blot analysis
- Harvested cells were lysed in Cell Lysis Buffer (Cell Signaling Technology, Beverly, MA, USA) for 20 minutes and centrifuged at 13,500 rpm for 5 minutes. Twenty micrograms of proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using 8%–12% gels and transferred to nitrocellulose membranes (Potran nitrocellulose membrane, Whatman, Kent, UK) using an iBlot dry blotting system (Invitrogen). The membranes were blocked with 5% non-fat dried milk and incubated with specific primary antibodies for (1) GADD34 (1:1,000), activating transcription factor 4 (ATF4; 1:1 000), p53 (1:1,000), and β-actin (1:2,000) from Santa Cruz Biotechnology (Dallas, TX, USA); (2) eIF2 (1:1,000), phospho-eIF2 (p-eIF2, 1:1,000), p-p53 (Ser15, 1:2,000), p21Waf1/Cip1 (1:500), cyclin B1 (1:1,000), cyclin E2 (1:1,000), p-cdc2 (p-cdk1, 1:2,000), Bax (1:1,000), Bcl-xL (1:2,000), procaspase-9 (1:2,000), and cleaved caspase-9 (1:1,000) from Cell Signaling Technology. After incubation with appropriate horseradish peroxidase-conjugated secondary IgG antibodies, bands were detected using ECL reagent (Amersham-GE Healthcare Life Sciences, Pittsburgh, PA, USA).
- Cell cycle assay
- The effect of the agent on the cell cycle of HCC cell lines was analyzed by flow cytometry using propidium iodide (PI) staining. Briefly, 106 cells were seeded in 60-mm dishes and incubated with the agent (30 μM) or phosphate-buffered saline (PBS) as a control for 24 hours at 37°C. After incubation, the cells were fixed with ice-cold 70% ethanol at 4°C overnight, washed twice with PBS, incubated with 10 μg/mL RNase A, and stained with 30 μg/mL PI. The stained cells were subsequently analyzed using flow cytometry (FACSCalibur machine, Becton Dickinson, Mountain View, CA, USA) with a 560-nm dichroic mirror and a 600-nm pass filter (bandwidth, 35 μm). The percentages of cells in G1, S, and G2 phases were determined using Cell Quest 3.1 software (Becton Dickinson).
MATERIALS AND METHODS
- GA screened as a potential therapeutic drug for HCC
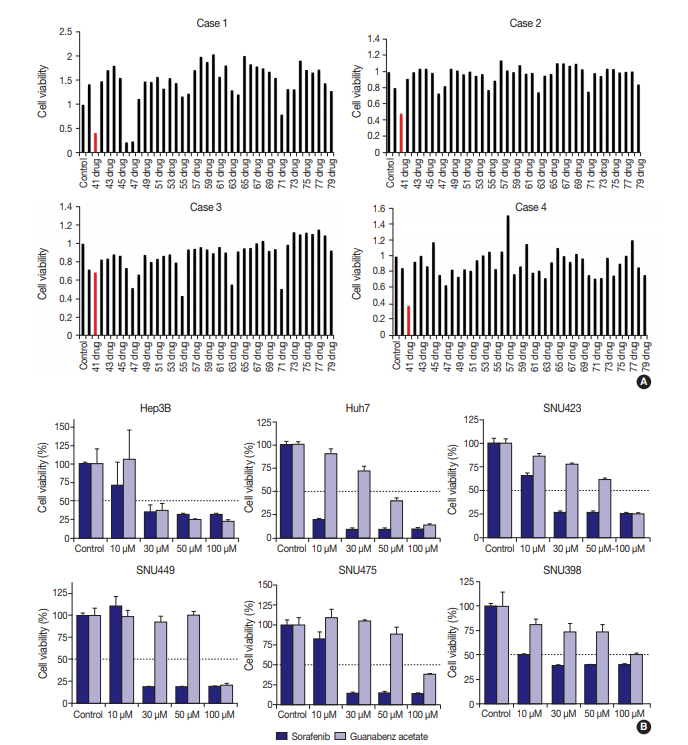
- Four primary cultured HCC cells were established using surgically resected fresh tumor tissues from four HCC patients and subsequently confirmed via pathologic examination (Fig. 1A, B). The clinicopathologic features of the patients are listed in Table 1. High-throughput drug screening using the Prestwick Chemical Library, which contains 1,120 FDA-approved drugs (Supplementary Table S1), was performed on the four primary cultured HCC cells. Two hundred and fifty-two of the 1,120 molecules were randomly selected and screened and identified GA as a candidate. The results were confirmed using individual in vitro assay. After 72-hour incubation with a concentration range of the molecules (0–50 μM), cell viability was inhibited more than 50% with 20 μM GA (i.e., compound 41) (Fig. 2A) in three out of four primary cultured HCC cells, measured using the MTT assay.
- GA inhibited growth of HCC cell lines
- To further validate the anti-cancer effect of GA in HCC, six HCC cell lines were treated with GA (Sigma-Aldrich) or with a control molecule, sorafenib. After 72-hour incubation with GA, growth of Hep3B (IC50 = 30 μM) and Huh7 (IC50 = 50 μM) were inhibited (Fig. 2B). The IC50 values of GA for SNU423, SNU449, and SNU475 cells were 100 μM, and the viability of SNU 398 cells did not drop below 50%, even at 100 μM GA (Fig. 2B). When the inhibitory effects of sorafenib on cell proliferation were compared with those of GA, the decline in viability occurred at similar or lower concentrations with sorafenib in all six HCC cell lines (Fig. 2B).
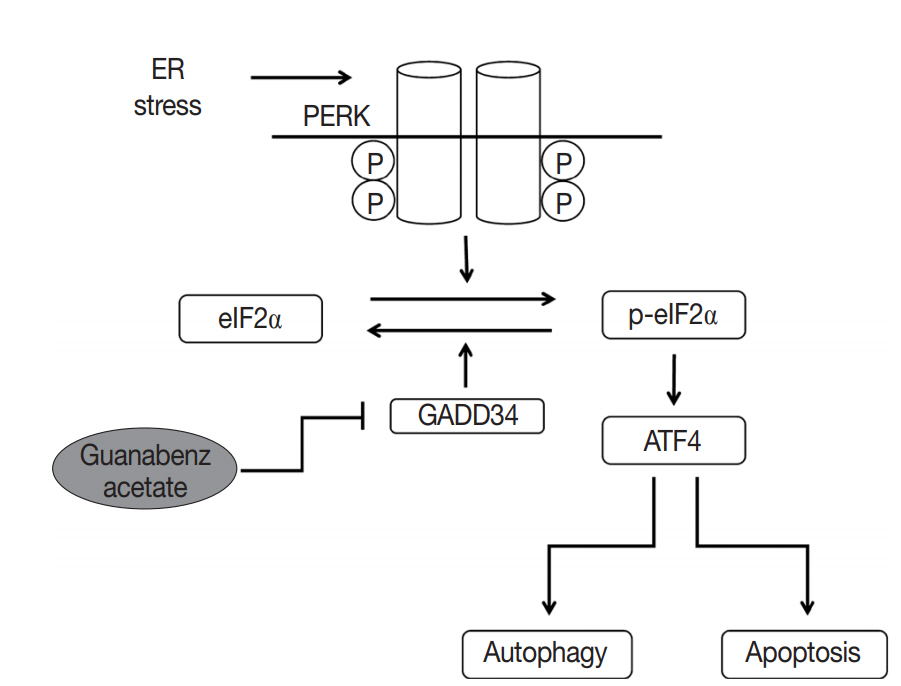
- GA targets PERK signaling pathway through GADD34 inactivation
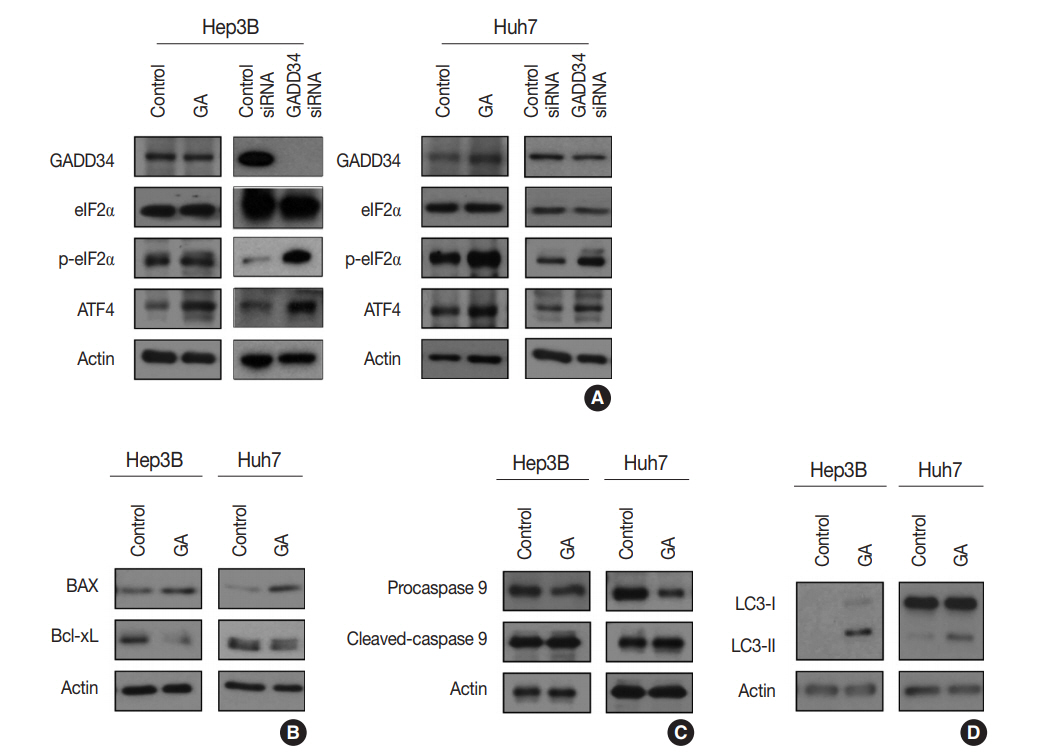
- GA is an agonist of the α2 adrenergic receptor that is used as an antihypertensive drug. GA is also known to block dephosphorylation of eIF2α by inactivation of GADD34 in the protein kinase RNA-like ER kinase (PERK) signaling pathway, which would finally lead to expression of genes involved in apoptosis and autophagy. Therefore, the protein expression levels of genes associated with PERK signaling were evaluated by western blotting in the Hep3B and Huh7 cell lines to study the mechanisms of GA in HCC. The level of p-eIF2α was upregulated, and ATF4 protein was increased after GA treatment of Hep3B and Huh7 cells (Fig. 3A). In addition, when HCC cells were transfected with GADD34 siRNA, levels of p-eIF2α and ATF4 were upregulated in both Hep3B and Huh7 HCC cells compared with levels in control HCC cells (Fig. 3A). These results reveal that GA acts as an inhibitor of dephosphorylation of eIF2α, likely by inactivation of GADD34 in HCC.
- GA induces apoptosis and autophagy-related cell death in HCC cells
- ATF4, a transcription factor that induces the expression of genes, is involved in amino acid responses, metabolism, antioxidant responses, apoptosis, autophagy, and GADD34-mediated effects. To assess if GA induces HCC cell death via apoptosis and/or autophagy following ATF4 upregulation, expression levels of apoptosis-related proteins (i.e., Bax, Bcl-xL, procaspase-9, and cleaved caspase-9) and an autophagy-related protein (i.e., microtubule-associated protein light chain 3, LC3) were examined in Hep3B and Huh7 by western blotting. In the Hep3B and Huh7 cells treated with GA, Bax protein expression was higher, while Bcl-xL protein expression was lower than the expressions in control HCC cells (Fig. 3B). In addition, protein levels of procaspase-9 were decreased, while cleaved caspase-9 was increased in both Hep3B and Huh7 cells (Fig. 3C). Both LC3-I and LC3-II protein levels increased in Hep3B, but only LC3-II protein level increased in Huh7 treated with GA (Fig. 3D). These results clearly demonstrate that both mechanism of apoptosis and autophagy play a role in GA-induced cell death. Activated ATF4 in the PERK pathway is considered an essential gateway for apoptosis and autophagy in HCC cells.
- Different types of cell cycle arrest in different HCC cell lines treated with GA
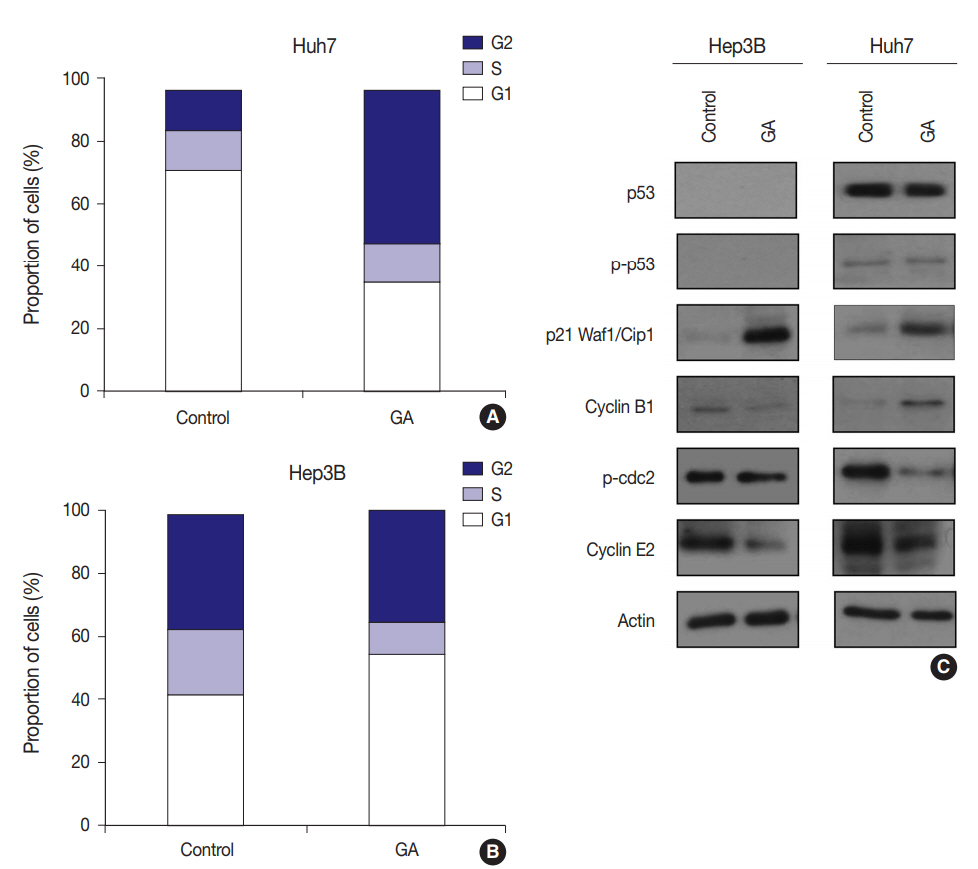
- To examine whether GA reduces HCC cell viability by inducing cell cycle arrest, flow cytometry was performed. The expression levels of checkpoint proteins in the G1/S and G2/M transitions and cell cycle regulating factors were then examined by western blotting. The cell cycle distribution pattern was different between the two HCC cell lines. In Hep3B cells treated with GA, the percentage of G1 phase cells increased, while the percentages of S and G2 phase cells concomitantly decreased (Fig. 4A). In contrast to Hep3B, the cell cycle distribution of Huh7 treated with GA revealed an increase in the percentage of G2 phase cells, while producing a concomitant decrease in the percentage of G1 phase cells (Fig. 4B). Among cell cycle-related proteins, the total protein level of p21 increased remarkably after GA treatment in Hep3B and Huh7 cells. The protein level of cyclin B1 slightly decreased, p-cdc2 remained unchanged, and cyclin E2 decreased in Hep3B cells. In Huh7 cells, however, the protein level of cyclin B1 increased, p-cdc2 remarkably decreased, and cyclin E2 slightly decreased (Fig. 4C). These results suggest that the reduced viability of HCC cells by GA treatment is regulated by cell cycle arrest. Moreover, the patterns of cell cycle arrest by GA treatment can vary among different HCC cell lines.
RESULTS
- In this study, we discovered that GA can be repurposed as a potential anti-cancer drug for HCC patients via preclinical drug efficacy screening by using primary cultured HCC cells that we established using the surgically resected HCCs and stable HCC cell lines.
- A new functional mechanism of GA was recently reported. In this mechanism, PERK activity, which is one of three pathways of the unfolded protein response (UPR) in the endoplasmic reticulum (ER), is increased by selective inactivation of GADD34-mediated dephosphorylation of eIF2α, subsequently inducing apoptosis and autophagy (summarized in Fig. 5) [21-24]. UPR acts as a protective mechanism to alleviate ER stress by increasing protein-folding capacity, inhibiting general protein translation, and promoting degradation of unfolded or misfolded proteins. Interestingly, the aggregation of unfolded or misfolded proteins resulting in ER stress is associated with progression of various diseases, such as cancer, metabolic diseases, diabetes, inflammation, liver dysfunction, neurodegenerative disorders, and brain and heart ischemia [22-25]. Moreover, when ER stress is persistent and cannot be resolved, apoptotic signaling is initiated, which eventually leads to cell death [23]. Thus, the UPR signaling pathway can be an attractive target for drug discovery in diverse diseases. Recent studies have demonstrated that GA acts as a novel therapeutic agent by inhibition of dephosphorylation of eIF2α in the UPR pathway for several disorders, including progressive neurodegenerative disease (especially amyotrophic lateral sclerosis), toxoplasmosis, and breast cancer [21,26-28].
- In the cases of HCC that were responsive to GA, the mechanisms of action were consistent with the known mechanism. First, we confirmed that GA acts on the PERK signaling pathway possibly by inactivation of GADD34 in HCC. The HCC cells treated with GA showed an increase in the phosphorylation of eIF2α and expression of ATF4. Moreover, both GA-treated HCC cells and GADD34 siRNA-transfected HCC cells showed the same patterns of p-eIF2α and ATF4, supporting that GA inhibits GADD34. Second, we also identified that GA reduced cell viability by both mechanisms of apoptosis and autophagy in HCC cells. In GA-treated HCC cells, apoptosis and autophagy reactions were confirmed by decreased expression of procaspase-9 and the increased levels of cleaved caspase-9 and LC3. We also discovered that GA independently affects the cell cycle. GA-treated HCC cells showed arrest in the G1 or G2 phase of the cell cycle, the pattern of which was different between the two HCC cell lines. Additional studies are needed to elucidate the reason why the phase of cell cycle arrest is different among HCC cell lines, and the mechanisms of these differences. In addition, the markedly upregulated p21Waf1/Cip1 protein levels in both Hep3B and Huh7 cells indicate that a p53-dependent pathway could be involved in the cell cycle arrest by GA.
- The present study is the first to perform a drug repurposing study using primary cultured cells that we established from HCC patients with a high-throughput screening approach to discover novel therapeutics. The drug discovery process was developed based on genome-based drug discovery, high-throughput screening, and combinatorial chemistry [17]. Contrary to expectations, novel agents identified through traditional methods have gradually declined because of high costs, time-consuming processes, and unexpected adverse reactions in clinical trials. This has led to a process called drug repositioning or repurposing [29,30]. Drug repositioning is a new approach for drug discovery and is believed to offer great benefits over the traditional method of searching for new active substances, since the safety and pharmacokinetics have already been established [17,18,31,32]. A representative new indication, introduced by drug repositioning, is thalidomide, a sedative hypnotic agent, which demonstrated significant clinical activity against multiple myeloma and leprosy [33,34]. Moreover, the combination therapy of drugs found by drug repositioning and conventional anticancer drugs may increase efficacy and reduce adverse reactions [17]. Thus, drug repositioning may become a key approach for treating variable malignancies, including HCC, in novel drug discovery.
- The commercially available libraries of marketed drugs and natural compounds have been widely used in drug screening tests for a broad spectrum of diseases, including cancers, viral or fungal infections, neurodegenerative disorders, and neuromuscular disorders, but never have been used for drug efficacy tests of HCC [35]. Although the unexpected effects of GA on the reduction of HCC cell viability were discovered from randomly selected drugs, the effects of GA on malignant cells have already been reported in breast cancer. A previous study revealed that combined treatment of GA and salubrinal can attenuate the malignant phenotype and tumor growth of triple negative breast cancer cells through the eIF2α-mediated Rac1 pathway [28]. As mentioned previously, GA acts as an inhibitor of dephosphorylation of eIF2α and can be used potentially as an anti-cancer drug for other malignancies, especially those originating from ER-rich organs.
- This study has a few limitations. It was performed in vitro and the GA IC50 value is high for a clinically achievable dose. Therefore, additional in vitro and in vivo studies are needed to conclusively determine whether GA can be a novel anti-cancer agent for inoperable HCC patients at a clinically achievable dose. In addition, molecular and immunohistochemical studies are needed to determine the predictive marker(s) for the therapeutic effects of GA in HCC patients. Finally, the effect of GA as an anti-cancer agent could be evaluated in other malignancies of ER-rich organs.
- In conclusion, GA, which was screened in primary cultured HCC cells, reduces HCC cell viability via the combined effects of apoptosis, autophagy, and cell cycle arrest. Therefore, GA may be repositioned as an anti-cancer therapeutic agent for HCC patients.
DISCUSSION
Electronic Supplementary Material
Author contributions
Conceptualization: HJK, HSS.
Data curation: HJK, HSS.
Formal analysis: HJK, HSS, SEL, YAS.
Methodology: HJK, HSS, SEL, YAS.
Supervision: EY, SJJ.
Visualization: HJK, HSS, YAS.
Writing—original draft: HJK.
Writing—review & editing: EY, SJJ, JK.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
- 1. Bosch FX, Ribes J, Cléries R, Díaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9: 191-211. ArticlePubMed
- 2. Shaw JJ, Shah SA. Rising incidence and demographics of hepatocellular carcinoma in the USA: what does it mean? Expert Rev Gastroenterol Hepatol 2011; 5: 365-70. ArticlePubMed
- 3. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362: 1907-17. ArticlePubMed
- 4. Karaman B, Battal B, Sari S, Verim S. Hepatocellular carcinoma review: current treatment, and evidence-based medicine. World J Gastroenterol 2014; 20: 18059-60. ArticlePubMedPMC
- 5. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012; 379: 1245-55. ArticlePubMed
- 6. Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist 2010; 15 Suppl 4: 5-13. ArticlePubMedPDF
- 7. Crissien AM, Frenette C. Current management of hepatocellular carcinoma. Gastroenterol Hepatol (N Y) 2014; 10: 153-61. PubMedPMC
- 8. Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs 2009; 69: 223-40. ArticlePubMed
- 9. Patel A, Sun W. Molecular targeted therapy in hepatocellular carcinoma: from biology to clinical practice and future. Curr Treat Options Oncol 2014; 15: 380-94. ArticlePubMedPDF
- 10. Berk V, Kaplan MA, Tonyali O, et al. Efficiency and side effects of sorafenib therapy for advanced hepatocellular carcinoma: a retrospective study by the anatolian society of medical oncology. Asian Pac J Cancer Prev 2013; 14: 7367-9. ArticlePubMedPDF
- 11. De Witt Hamer PC, Van Tilborg AA, Eijk PP, et al. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008; 27: 2091-6. ArticlePubMedPDF
- 12. Seol HS, Suh YA, Ryu YJ, et al. A patient-derived xenograft mouse model generated from primary cultured cells recapitulates patient tumors phenotypically and genetically. J Cancer Res Clin Oncol 2013; 139: 1471-80. ArticlePubMedPDF
- 13. Seol HS, Kang HJ, Lee SI, et al. Development and characterization of a colon PDX model that reproduces drug responsiveness and the mutation profiles of its original tumor. Cancer Lett 2014; 345: 56-64. ArticlePubMed
- 14. Mitra A, Mishra L, Li S. Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol 2013; 31: 347-54. ArticlePubMedPMC
- 15. Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci 2014; 10: 654-63. ArticlePubMedPMC
- 16. Würth R, Thellung S, Bajetto A, Mazzanti M, Florio T, Barbieri F. Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today 2016; 21: 190-9. ArticlePubMed
- 17. Banno K, Iida M, Yanokura M, et al. Drug repositioning for gynecologic tumors: a new therapeutic strategy for cancer. Scientific-WorldJournal 2015; 2015: 341362.ArticlePDF
- 18. Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, Schutjens MH. Drug repositioning and repurposing: terminology and definitions in literature. Drug Discov Today 2015; 20: 1027-34. ArticlePubMed
- 19. Saxena A, Becker D, Preeshagul I, Lee K, Katz E, Levy B. Therapeutic effects of repurposed therapies in non-small cell lung cancer: what is old is new again. Oncologist 2015; 20: 934-45. ArticlePubMedPMCPDF
- 20. Oprea TI, Overington JP. Computational and practical aspects of drug repositioning. Assay Drug Dev Technol 2015; 13: 299-306. ArticlePubMedPMC
- 21. Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis 2014; 71: 317-24. ArticlePubMed
- 22. Rozpedek W, Markiewicz L, Diehl JA, Pytel D, Majsterek I. Unfolded protein response and PERK kinase as a new therapeutic target in the pathogenesis of Alzheimer's disease. Curr Med Chem 2015; 22: 3169-84. ArticlePubMedPMC
- 23. Shah SZ, Zhao D, Khan SH, Yang L. Unfolded protein response pathways in neurodegenerative diseases. J Mol Neurosci 2015; 57: 529-37. ArticlePubMedPDF
- 24. Stone S, Lin W. The unfolded protein response in multiple sclerosis. Front Neurosci 2015; 9: 264.ArticlePubMedPMC
- 25. Harada M, Nose E, Takahashi N, et al. Evidence of the activation of unfolded protein response in granulosa and cumulus cells during follicular growth and maturation. Gynecol Endocrinol 2015; 31: 783-7. ArticlePubMed
- 26. Vieira FG, Ping Q, Moreno AJ, et al. Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS One 2015; 10: e0135570.ArticlePubMedPMC
- 27. Benmerzouga I, Checkley LA, Ferdig MT, Arrizabalaga G, Wek RC, Sullivan WJ Jr. Guanabenz repurposed as an antiparasitic with activity against acute and latent toxoplasmosis. Antimicrob Agents Chemother 2015; 59: 6939-45. ArticlePubMedPMCPDF
- 28. Hamamura K, Minami K, Tanjung N, et al. Attenuation of malignant phenotypes of breast cancer cells through eIF2alpha-mediated downregulation of Rac1 signaling. Int J Oncol 2014; 44: 1980-8. PubMed
- 29. Jadamba E, Shin M. A systematic framework for drug repositioning from integrated omics and drug phenotype profiles using pathway-drug network. Biomed Res Int 2016; 2016: 7147039.ArticlePubMedPMCPDF
- 30. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 2004; 3: 673-83. ArticlePubMedPDF
- 31. Temesi G, Bolgár B, Arany A, Szalai C, Antal P, Mátyus P. Early repositioning through compound set enrichment analysis: a knowledgerecycling strategy. Future Med Chem 2014; 6: 563-75. ArticlePubMed
- 32. Mizushima T. Identification of a molecular mechanism for actions of existing medicines and its application for drug development. Yakugaku Zasshi 2012; 132: 713-20. ArticlePubMed
- 33. Schwab C, Jagannath S. The role of thalidomide in multiple myeloma. Clin Lymphoma Myeloma 2006; 7: 26-9. ArticlePubMed
- 34. Walker SL, Waters MF, Lockwood DN. The role of thalidomide in the management of erythema nodosum leprosum. Lepr Rev 2007; 78: 197-215. ArticlePubMed
- 35. Choulier L, Nomine Y, Zeder-Lutz G, et al. Chemical library screening using a SPR-based inhibition in solution assay: simulations and experimental validation. Anal Chem 2013; 85: 8787-95. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- ER stress signaling at the interphase between MASH and HCC
Younis Hazari, Eric Chevet, Béatrice Bailly-Maitre, Claudio Hetz
Hepatology.2026; 83(2): 387. CrossRef - The Integrated Stress Response in Cancer: Paradox and Therapeutic Promise
Chenliang Zhang, Ting Zhang, Qiulin Tang, Yu Zeng, Dan Cao
Comprehensive Physiology.2026;[Epub] CrossRef - Optogenetic Control of the Integrated Stress Response Limits Glioblastoma Invasion
Lisa K. Månsson, Ethan Dickson, Lun Hao, Angela A. Pitenis, Maxwell Z. Wilson
Cell Biochemistry and Function.2026;[Epub] CrossRef - The Construction of ceRNA Regulatory Network Unraveled Prognostic Biomarkers and Repositioned Drug Candidates for the Management of Pancreatic Ductal Adenocarcinoma
Busra Aydin, Keziban Okutan, Ozge Onluturk Aydogan, Raghu Sinha, Beste Turanli
Current Issues in Molecular Biology.2025; 47(7): 496. CrossRef - Current trends and future prospects of drug repositioning in gastrointestinal oncology
Nayeralsadat Fatemi, Mina Karimpour, Hoda Bahrami, Mohammad Reza Zali, Vahid Chaleshi, Andrea Riccio, Ehsan Nazemalhosseini-Mojarad, Mehdi Totonchi
Frontiers in Pharmacology.2024;[Epub] CrossRef - Small molecules for impairing endoplasmic reticulum in cancer
Tripti Mishra, Navneet Dubey, Sudipta Basu
Organic & Biomolecular Chemistry.2024; 22(44): 8689. CrossRef -
Guanabenz acetate, an antihypertensive drug repurposed as an inhibitor of
Escherichia coli
biofilm
Arakkaveettil Kabeer Farha, Olivier Habimana, Harold Corke, Olaya Rendueles
Microbiology Spectrum.2024;[Epub] CrossRef - The integrated stress response in cancer progression: a force for plasticity and resistance
Caleb L. Lines, Morgan J. McGrath, Tanis Dorwart, Crystal S. Conn
Frontiers in Oncology.2023;[Epub] CrossRef - Endoplasmic reticulum stress: Multiple regulatory roles in hepatocellular carcinoma
Jiacheng Wu, Shan Qiao, Yien Xiang, Menying Cui, Xiaoxiao Yao, Ruixin Lin, Xuewen Zhang
Biomedicine & Pharmacotherapy.2021; 142: 112005. CrossRef - The two faces of the Integrated Stress Response in cancer progression and therapeutic strategies
Eugenia Licari, Luis Sánchez-del-Campo, Paola Falletta
The International Journal of Biochemistry & Cell Biology.2021; 139: 106059. CrossRef - Repurposing of Guanabenz acetate by encapsulation into long-circulating nanopolymersomes for treatment of triple-negative breast cancer
Yusuf A. Haggag, Mohamed Yasser, Murtaza M. Tambuwala, Suleiman S. El Tokhy, Mohammad Isreb, Ahmed A. Donia
International Journal of Pharmaceutics.2021; 600: 120532. CrossRef - Endoplasmic reticulum stress: New insights into the pathogenesis and treatment of retinal degenerative diseases
Marina S. Gorbatyuk, Christopher R. Starr, Oleg S. Gorbatyuk
Progress in Retinal and Eye Research.2020; 79: 100860. CrossRef - Delineating the role of eIF2α in retinal degeneration
Christopher R. Starr, Marina S. Gorbatyuk
Cell Death & Disease.2019;[Epub] CrossRef - Repositioning of Guanabenz in Conjugation with Gold and Silver Nanoparticles against Pathogenic Amoebae Acanthamoeba castellanii and Naegleria fowleri
Areeba Anwar, Mohammad Ridwane Mungroo, Ayaz Anwar, William J. Sullivan, Naveed Ahmed Khan, Ruqaiyyah Siddiqui
ACS Infectious Diseases.2019; 5(12): 2039. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
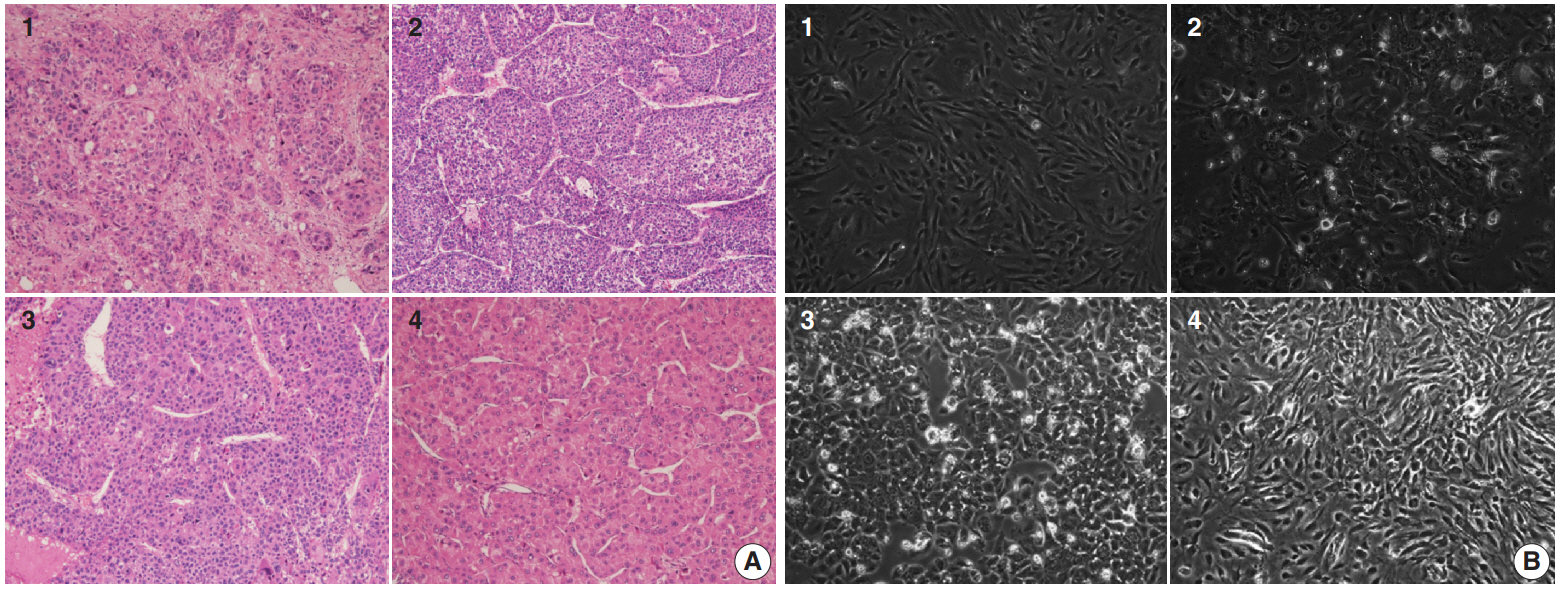
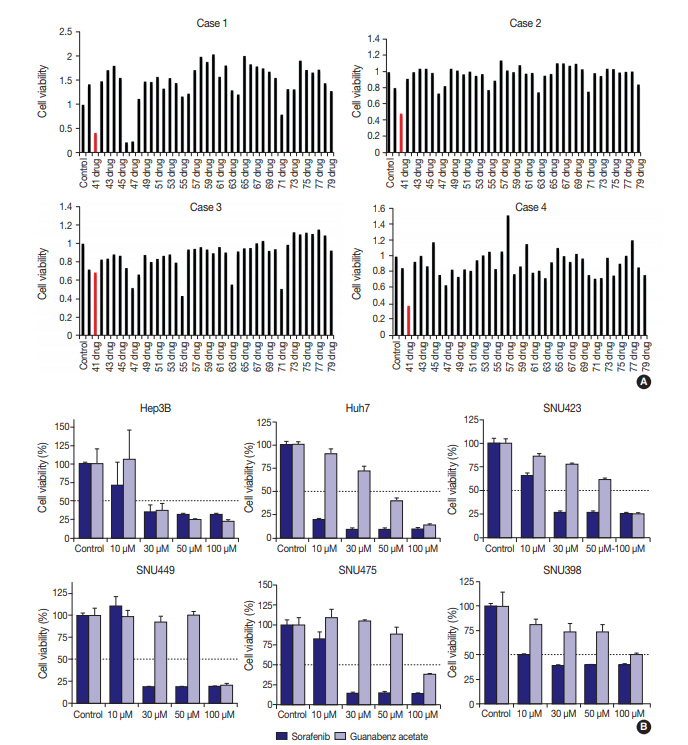
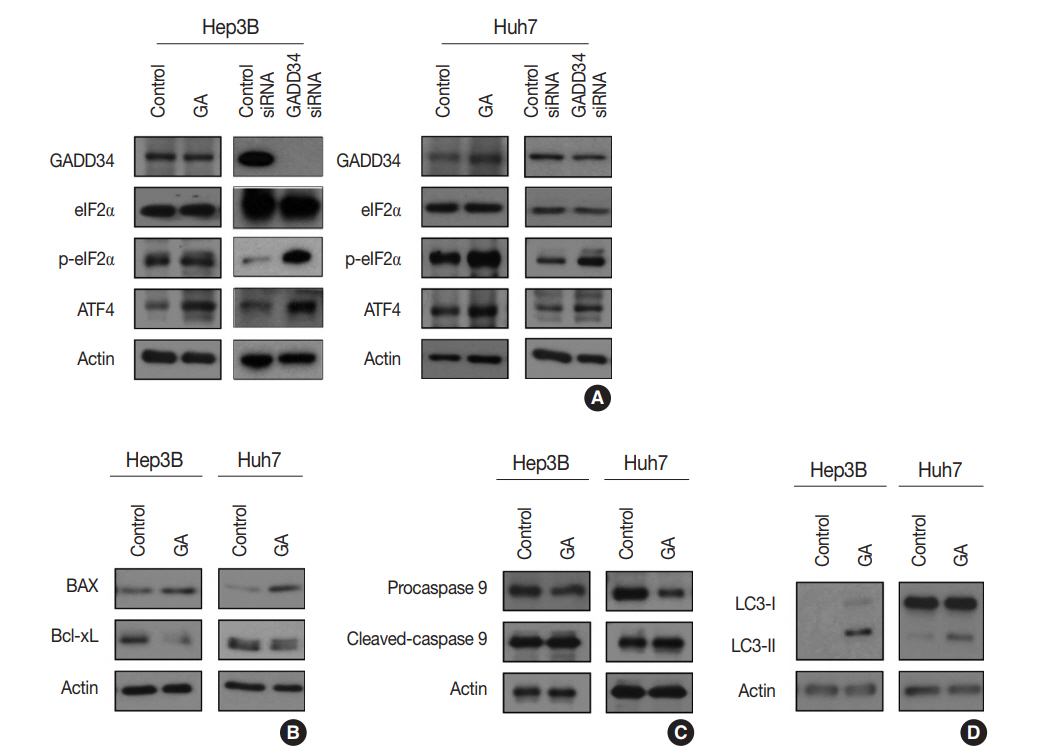
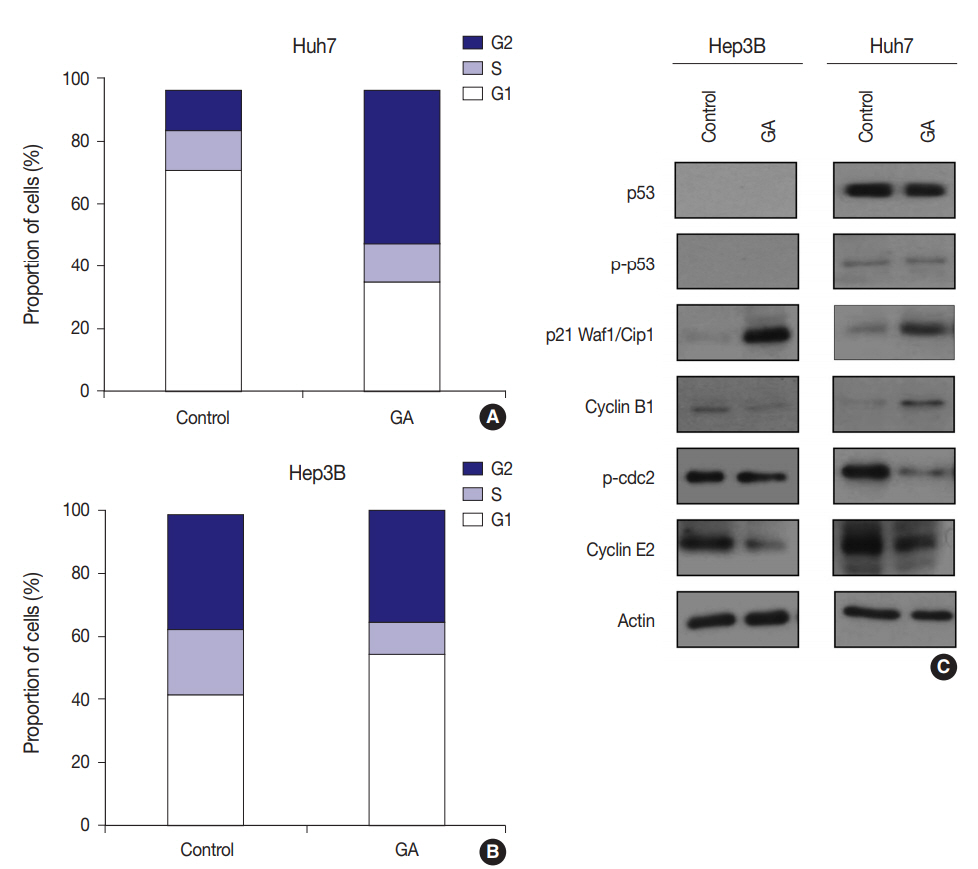
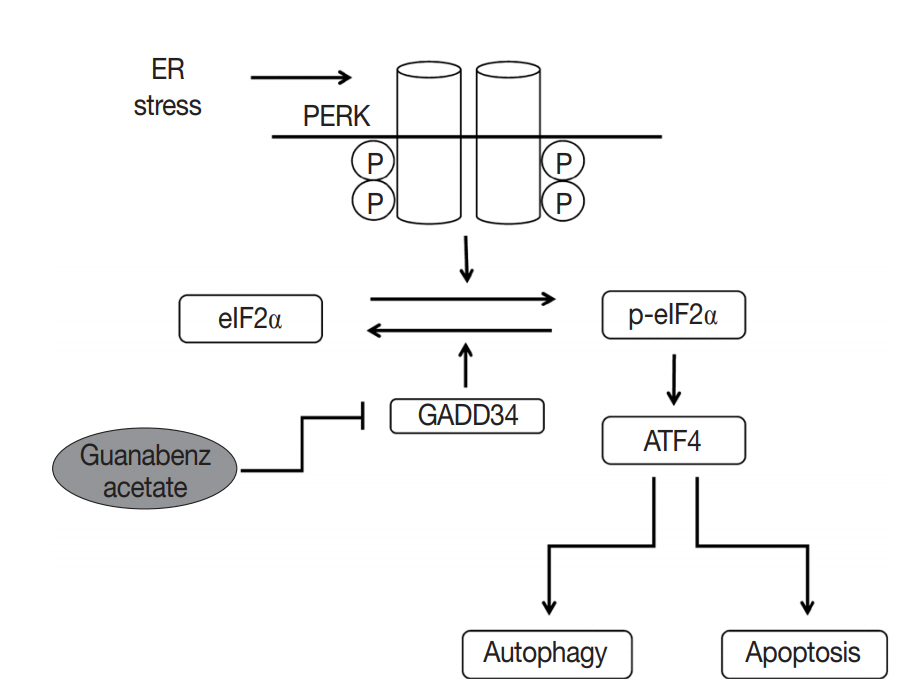





Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
| Case No. | Age (yr) | Sex | Size (cm) | Edmondson-Steiner grade | Underlying disease | Previous treatment |
|---|---|---|---|---|---|---|
| 1 | 58 | M | 13 | III | Cirrhosis/HBV | No |
| 2 | 56 | M | 4.5 | III | Periportal fibrosis/HBV | No |
| 3 | 70 | M | 4.7 | III | Cirrhosis/HBV | TACE |
| 4 | 55 | M | 11 | II | Cirrhosis/HBV | No |
PDX, patient derived xenograft; HBV, hepatitis B virus; TACE, transarterial chemoembolization.

