 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 50(6); 2016 > Article
-
Review
Pathogenesis of Focal Segmental Glomerulosclerosis - Beom Jin Lim, Jae Won Yang1, Woo Sung Do, Agnes B. Fogo2
-
Journal of Pathology and Translational Medicine 2016;50(6):405-410.
DOI: https://doi.org/10.4132/jptm.2016.09.21
Published online: October 16, 2016
Department of Pathology, Yonsei University College of Medicine, Seoul, Korea
1Department of Nephrology, Yonsei University Wonju College of Medicine, Wonju, Korea
2Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, USA
- Corresponding Author Agnes B. Fogo, MD MCN C3310, Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA Tel: +1-615-322-3114 Fax: +1-615-343-7023 E-mail: agnes.fogo@vanderbilt.edu
• Received: August 23, 2016 • Accepted: September 21, 2016
© 2016 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Focal segmental glomerulosclerosis (FSGS) is characterized by focal and segmental obliteration of glomerular capillary tufts with increased matrix. FSGS is classified as collapsing, tip, cellular, perihilar and not otherwise specified variants according to the location and character of the sclerotic lesion. Primary or idiopathic FSGS is considered to be related to podocyte injury, and the pathogenesis of podocyte injury has been actively investigated. Several circulating factors affecting podocyte permeability barrier have been proposed, but not proven to cause FSGS. FSGS may also be caused by genetic alterations. These genes are mainly those regulating slit diaphragm structure, actin cytoskeleton of podocytes, and foot process structure. The mode of inheritance and age of onset are different according to the gene involved. Recently, the role of parietal epithelial cells (PECs) has been highlighted. Podocytes and PECs have common mesenchymal progenitors, therefore, PECs could be a source of podocyte repopulation after podocyte injury. Activated PECs migrate along adhesion to the glomerular tuft and may also contribute to the progression of sclerosis. Markers of activated PECs, including CD44, could be used to distinguish FSGS from minimal change disease. The pathogenesis of FSGS is very complex; however, understanding basic mechanisms of podocyte injury is important not only for basic research, but also for daily diagnostic pathology practice.
- There are several observations indicating that podocyte injury is at the center of the development of FSGS. First, podocyte injury is the earliest morphologic feature of FSGS. In recurrent FSGS in the allograft kidney, podocyte injury is detected by electron microscopy prior to the development of overt sclerosis [14-16]. Second, there are animal models of podocyte-specific injury resulting in FSGS. NEP25 mice express human CD25 specifically on podocytes. Injection of immunotoxin which binds to human CD25 induced podocyte-specific injury and FSGS occurred a few weeks later [17]. Rats expressing diphtheria toxin receptors on podocytes developed FSGS after diphtheria toxin injection [18]. Third, histologic appearance of FSGS and clinical symptoms were in proportion with the number of injured podocytes [18]. Therefore, the pathogenesis of podocyte injury is a key to understand the characteristics of FSGS.
PATHOGENESIS OF PODOCYTE INJURY
- As for the pathophysiology of podocyte injury, several mechanisms have been proposed with supporting evidences. Circulating permeability factors have been reckoned as the initiating factor of podocyte injury in primary FSGS and its recurrence after transplantation. The presence of serum factors that can cause podocyte injury was suggested from the therapeutic effect of immunoadsorption therapy [19] and observations that plasmapheresis could decrease the glomerular injury induced by patients’ serum [20]. Further, serum of recurrent FSGS patients significantly increased albumin permeability of glomeruli in an in vitro test [21]. Among the proposed circulating permeability factors, soluble urokinase receptor (suPAR) has been most thoroughly investigated. Wei et al. [22] presented data in a mouse model suggesting that the urokinase receptor of podocytes contributed to podocyte loss and proteinuria. The same group also suggested that suPAR could be the cause of FSGS. Serum levels of suPAR were increased in about two-thirds of primary FSGS patients and were also associated with recurrent FSGS after transplantation [23]. They also demonstrated that suPAR was increased in two different cohorts of biopsyproven FSGS patients. However, suPAR was inversely correlated with estimated glomerular filtration rate (eGFR) and treatment response [24]. Other authors found that plasma suPAR level was significantly increased in FSGS patients versus patients with minimal change disease, membranous nephropathy, or normal control. However, suPAR level was not useful in distinguishing primary and secondary FSGS [25]. Several contradicting reports on suPAR have also been published. Importantly, eGFR affects plasma levels of suPAR in patients with non-FSGS glomerular lesions, and a suPAR cut off value could not be determined even in FSGS patients due to the effect of eGFR [26]. Plasma levels of suPAR were also increased in lupus nephritis patients compared to lupus patients without renal involvement [27]. In IgA nephropathy patients, the plasma level of suPAR was related to the development of secondary segmental sclerosis [28,29]. Therefore, whether suPAR plays a role in the development of focal segmental lesions and its specificity to the primary FSGS are still open to further investigation.
- Cardiotrophin-like cytokine-1 (CLC-1 or cardiotrophin-like cytokine factor 1 [CLCF-1]) is another candidate circulating permeability factor for primary FSGS. Savin et al. [30] have published on a serum factor purified from FSGS patients, which increased albumin permeability in isolated rat glomeruli. This factor had affinity for galactose and its molecular weight was less than 30 kDa. They identified this factor as CLC-1 by proteomic analysis and also found that the activity of CLC-1 was decreased by several factors such as heterodimer formation with cosecreted cytokine receptor-like factor 1 (CRLF1), Janus kinase 2 (JAK2) inhibitor, and signal transducer and activator of transcription 3 (STAT3) inhibitor [31,32]. A phase II clinical trial on therapeutic effect of galactose in patients with steroid-resistant FSGS was performed with inconclusive results due to small sample size [33,34]. This study design is interesting, considering that CLC-1 has high affinity for galactose and the CLC-1–galacotse complex can be easily removed in the liver [30].
- Though known to be related to minimal change disease rather than FSGS, angiopoietin-like-4 (Angptl4) is also of interest [35]. Angptl4 has different functions according to its sialylation. While proteinuria was induced by hyposialylated Angptl4 located within the glomerulus, normosialylated Angptl4 was present in the peripheral circulation and mediated hypertriglyceridemia [35,36], indicating that these two symptoms of nephrotic syndrome could be linked through a common circulating factor.
CIRCULATING PERMEABILITY FACTORS
- FSGS, as a podocytopathy, may be caused by mutation in several genes, which are important in maintaining podocyte morphology and function. Most of these genes can be categorized as those which are related with slit diaphragm structure, actin cytoskeleton of podocytes, or podocyte-glomerular basement membrane interaction through foot processes [37-39]. In addition, a specific channel mutation (see below) has also been identified as a cause of FSGS (Table 1). Alteration of these genes results in autosomal dominant or recessive congenital, infantile, or late onset nephrotic syndrome, some of which presents as FSGS histologically [40]. Mutation in the NPHS1 gene and resulting loss of its product nephrin, are responsible for congenital nephrotic syndrome of Finnish type. The locus of NPHS1 was identified at 19q13.1 in 1998 [41], which was the first identification of a podocytopathy-related gene. After this discovery, NPHS2 [42,43], PLCE1 (phospholipase Cε1, NPHS3) [43], WT1 (Wilms tumor 1, NPHS4) [44], LAMB2 (laminin β2, NPHS5) [45], PTPRO (protein tyrosine phosphatase receptor type O, NPHS6) [46], ARHGDIA (Rho GDP dissociation inhibitor α, NPHS8) [47], ADCK4 (aarF domain containing kinase 4, NPHS9) [48], and EMP2 (epithelial membrane protein 2, NPHS10) [49] were identified and related to autosomal recessive nephrotic syndrome. Many other genes related to nephrotic syndrome have been identified including ACTN4 (actinin α4, FSGS1) [50], TRPC6 (transient receptor potential cation channel 6, FSGS2) [51], CD2AP (CD2-associated protein, FSGS3) [52], APOL1 (apolipoprotein L1, FSGS4) [53], INF2 (inverted formin, FSGS5) [54], MYO1E (myosin 1E, FSGS6) [55], PAX2 (paired box gene 2, FSGS7) [56], ANLN (anillin, FSGS8) [57], and CRB2 (Crumbs homolog 2, FSGS9) [58]. There are interactions of these genes and their products. For example, WT1 transcriptionally regulates nephrin encoding of NPHS1, therefore, WT1 mutations influence NPHS1 function [59]. A study in a European cohort reported that two thirds of nephrotic syndrome within 1 year of life are related to alteration of NPHS1, NPHS2, WT1, or LAMB2 [60]. Another study in a non-Finnish ethnic group also reported that NPHS1 and NPHS2 mutations were the most common genetic alterations in congenital nephrotic syndrome [61]. In contrast to these Western studies, a genetic analysis of 30 Korean congenital and infantile nephrotic syndrome patients revealed that WT1 and NPHS1 mutations were the most frequent alterations, while NPHS2 mutations were the lowest frequency genetic alteration [62].
GENETIC BACKGROUND
- Podocytes are terminally differentiated cells having very limited ability of regeneration or proliferation. Therefore, the mechanism of repopulation of podocytes after podocyte injury has been of great interest. Recently, it has been suggested that parietal epithelial cells (PECs) lining Bowman’s capsule play an important role in this process by migrating from their original site to replace injured podocytes [63]. During glomerulogenesis, PECs and podocytes originate from common mesenchymal progenitors and finally have different phenotypes. Although little is known about the function of terminally differentiated PECs, they express tight junction molecules such as claudin-1, zonula occludens-1, and occludin and have barrier function against protein [64]. Some PECs express both CD133 and CD24, which are known to be stem cell markers, and these cells have regenerative ability [65]. More detailed study revealed that PECs show hierarchical differentiation according to their locations. PECs located at the urinary pole express CD133 and CD24 without the expression of podocyte markers (nestin, complement receptor-1, and podocalyxin). PECs of the vascular pole express podocyte markers without the expression of CD133 or CD24. In other areas, PECs express both CD133/CD24 and podocyte markers [66]. CD133 and CD24-expressing PECs have the ability to ameliorate kidney injury by potentiating tubular regeneration [65] and podocyte replacement, however, they can also contribute to glomerular injury such as glomerulosclerosis and crescent formation [67,68]. Animal models and human posttransplant biopsies demonstrated that invasion of activated PECs through the adhesion sites of the capillary tuft contributed to the development of FSGS [69]. The adhesion of the glomerular tuft to the Bowman’s capsule as a bridge of PEC migration appears to occur at early stages of FSGS development [70]. Therefore, detecting activated PECs on Bowman’s capsule or on the glomerular tuft could be an adjunctive diagnostic tool for early FSGS. In support of this concept, CD44 as a marker of activated PECs successfully distinguished early primary FSGS [70] and early post-transplant recurrence of FSGS [71] from minimal change disease. Interestingly, mutation of ARHGDIA, which is responsible for nephrotic syndrome, increased migration activity of cultured podocytes [47].
PARIETAL EPITHELIAL CELLS AND PODOCYTE INJURY
- The etiology and pathogenesis of FSGS are very complex. Current research is focusing on the role of podocytes and interaction with PECs. Understanding the mechanism of podocyte injury, its progression and possible recovery is important not only for basic research but also for daily diagnostic pathology practice.
CONCLUSION
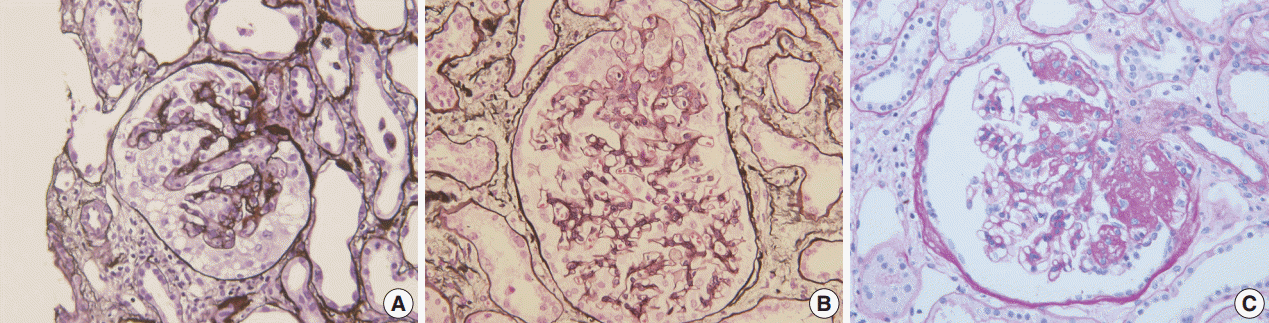
Fig. 1.Morphologic variants of focal segmental glomerulosclerosis. (A) Capillary collapse and podocyte hyperplasia are characteristic features of the collapsing variant. (B) In the tip variant, segmental lesion involves the glomerular tuft next to the tubular pole. (C) The perihilar variant is diagnosed when sclerosis or hyalinosis are present in perihilar lesion in more than half of the sclerotic glomeruli.
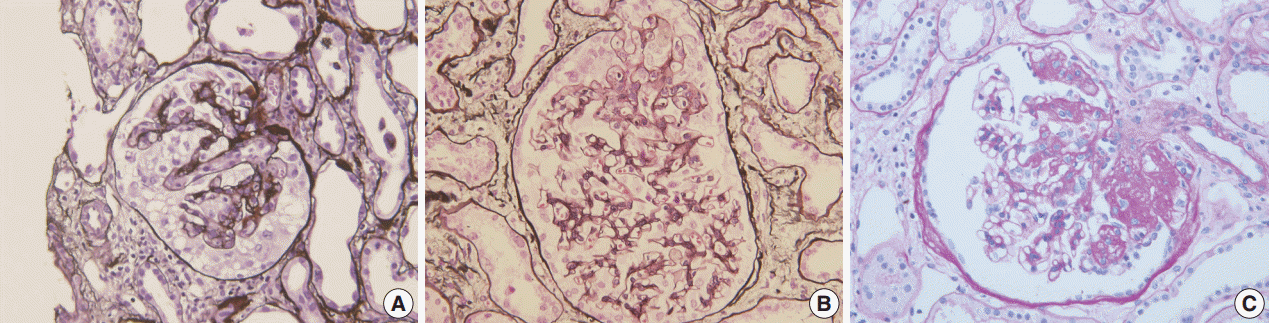
Table 1.Genes related to FSGS or nephrotic syndrome
- 1. D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 2004; 43: 368-82. ArticlePubMed
- 2. Weiss MA, Daquioag E, Margolin EG, Pollak VE. Nephrotic syndrome, progressive irreversible renal failure, and glomerular “collapse”: a new clinicopathologic entity? Am J Kidney Dis 1986; 7: 20-8. ArticlePubMed
- 3. Moudgil A, Nast CC, Bagga A, et al. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int 2001; 59: 2126-33. ArticlePubMed
- 4. Markowitz GS, Appel GB, Fine PL, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol 2001; 12: 1164-72. ArticlePubMed
- 5. Markowitz GS, Nasr SH, Stokes MB, D’Agati VD. Treatment with IFN-α, -β, or -γ is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2010; 5: 607-15. ArticlePubMedPMC
- 6. Mohamed N, Goldstein J, Schiff J, John R. Collapsing glomerulopathy following anthracycline therapy. Am J Kidney Dis 2013; 61: 778-81. ArticlePubMed
- 7. Gulati A, Sharma A, Hari P, Dinda AK, Bagga A. Idiopathic collapsing glomerulopathy in children. Clin Exp Nephrol 2008; 12: 348-53. ArticlePubMedPDF
- 8. D’Agati VD, Alster JM, Jennette JC, et al. Association of histologic variants in FSGS clinical trial with presenting features and outcomes. Clin J Am Soc Nephrol 2013; 8: 399-406. ArticlePubMed
- 9. Trivedi M, Pasari A, Chowdhury AR, Abraham A, Pandey R. Tip variant of focal segmental glomerulosclerosis: is it truly a benign variant? Ren Fail 2015; 37: 763-8. ArticlePubMed
- 10. Kwon YE, Han SH, Kie JH, et al. Clinical features and outcomes of focal segmental glomerulosclerosis pathologic variants in Korean adult patients. BMC Nephrol 2014; 15: 52.ArticlePubMedPMCPDF
- 11. Paik KH, Lee BH, Cho HY, et al. Primary focal segmental glomerular sclerosis in children: clinical course and prognosis. Pediatr Nephrol 2007; 22: 389-95. ArticlePubMedPDF
- 12. Praditpornsilpa K, Napathorn S, Yenrudi S, Wankrairot P, Tungsaga K, Sitprija V. Renal pathology and HIV infection in Thailand. Am J Kidney Dis 1999; 33: 282-6. ArticlePubMed
- 13. Naaz I, Wani R, Najar MS, Banday K, Baba KM, Jeelani H. Collapsing glomerulopathy in an HIV-positive patient in a low-incidence belt. Indian J Nephrol 2010; 20: 211-3. ArticlePubMedPMC
- 14. Chang JW, Pardo V, Sageshima J, et al. Podocyte foot process effacement in postreperfusion allograft biopsies correlates with early recurrence of proteinuria in focal segmental glomerulosclerosis. Transplantation 2012; 93: 1238-44. ArticlePubMedPMC
- 15. Alachkar N, Wei C, Arend LJ, et al. Podocyte effacement closely links to suPAR levels at time of posttransplantation focal segmental glomerulosclerosis occurrence and improves with therapy. Transplantation 2013; 96: 649-56. ArticlePubMedPMC
- 16. Canaud G, Dion D, Zuber J, et al. Recurrence of nephrotic syndrome after transplantation in a mixed population of children and adults: course of glomerular lesions and value of the Columbia classification of histological variants of focal and segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant 2010; 25: 1321-8. ArticlePubMed
- 17. Matsusaka T, Xin J, Niwa S, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol 2005; 16: 1013-23. ArticlePubMed
- 18. Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 2005; 16: 2941-52. PubMed
- 19. Haas M, Godfrin Y, Oberbauer R, et al. Plasma immunadsorption treatment in patients with primary focal and segmental glomerulosclerosis. Nephrol Dial Transplant 1998; 13: 2013-6. ArticlePubMed
- 20. Artero ML, Sharma R, Savin VJ, Vincenti F. Plasmapheresis reduces proteinuria and serum capacity to injure glomeruli in patients with recurrent focal glomerulosclerosis. Am J Kidney Dis 1994; 23: 574-81. ArticlePubMed
- 21. Savin VJ, Sharma R, Sharma M, et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med 1996; 334: 878-83. ArticlePubMed
- 22. Wei C, Moller CC, Altintas MM, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med 2008; 14: 55-63. ArticlePubMedPDF
- 23. Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 2011; 17: 952-60. PubMedPMC
- 24. Wei C, Trachtman H, Li J, et al. Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol 2012; 23: 2051-9. ArticlePubMedPMC
- 25. Huang J, Liu G, Zhang YM, et al. Plasma soluble urokinase receptor levels are increased but do not distinguish primary from secondary focal segmental glomerulosclerosis. Kidney Int 2013; 84: 366-72. ArticlePubMed
- 26. Meijers B, Maas RJ, Sprangers B, et al. The soluble urokinase receptor is not a clinical marker for focal segmental glomerulosclerosis. Kidney Int 2014; 85: 636-40. ArticlePubMed
- 27. Qin DD, Song D, Huang J, Yu F, Zhao MH. Plasma-soluble urokinase-type plasminogen activator receptor levels are associated with clinical and pathological activities in lupus nephritis: a large cohort study from China. Lupus 2015; 24: 546-57. ArticlePubMedPDF
- 28. Guo SM, Han M, Chen MX, et al. Soluble urokinase receptor levels are correlated with focal segmental glomerulosclerosis lesions in IgA nephropathy: a cohort study from China. PLoS One 2015; 10: e0138718. Article
- 29. Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Cattran DC, Coppo R, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 2009; 76: 534-45. PubMed
- 30. Savin VJ, McCarthy ET, Sharma R, Charba D, Sharma M. Galactose binds to focal segmental glomerulosclerosis permeability factor and inhibits its activity. Transl Res 2008; 151: 288-92. ArticlePubMed
- 31. McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2010; 5: 2115-21. ArticlePubMed
- 32. Sharma M, Zhou J, Gauchat JF, et al. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl Res 2015; 166: 384-98. ArticlePubMedPMC
- 33. Trachtman H, Vento S, Gipson D, et al. Novel therapies for resistant focal segmental glomerulosclerosis (FONT) phase II clinical trial: study design. BMC Nephrol 2011; 12: 8.ArticlePubMedPMCPDF
- 34. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol 2015; 16: 111.ArticlePubMedPMCPDF
- 35. Clement LC, Avila-Casado C, Macé C, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med 2011; 17: 117-22. ArticlePubMedPDF
- 36. Clement LC, Macé C, Avila-Casado C, Joles JA, Kersten S, Chugh SS. Circulating angiopoietin-like 4 links proteinuria with hypertriglyceridemia in nephrotic syndrome. Nat Med 2014; 20: 37-46. ArticlePubMedPDF
- 37. Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol 2015; 16: 101.ArticlePubMedPMCPDF
- 38. Jefferson JA, Shankland SJ. The pathogenesis of focal segmental glomerulosclerosis. Adv Chronic Kidney Dis 2014; 21: 408-16. ArticlePubMedPMC
- 39. D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med 2011; 365: 2398-411. ArticlePubMed
- 40. Pollak MR. Familial FSGS. Adv Chronic Kidney Dis 2014; 21: 422-5. ArticlePubMedPMC
- 41. Kestilä M, Lenkkeri U, Männikkö M, et al. Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome. Mol Cell 1998; 1: 575-82. ArticlePubMed
- 42. Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000; 24: 349-54. ArticlePubMedPDF
- 43. Hinkes B, Wiggins RC, Gbadegesin R, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006; 38: 1397-405. PubMed
- 44. Mucha B, Ozaltin F, Hinkes BG, et al. Mutations in the Wilms' tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr Res 2006; 59: 325-31. ArticlePubMed
- 45. Zenker M, Aigner T, Wendler O, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 2004; 13: 2625-32. PubMed
- 46. Ozaltin F, Ibsirlioglu T, Taskiran EZ, et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am J Hum Genet 2011; 89: 139-47. ArticlePubMedPMC
- 47. Gee HY, Saisawat P, Ashraf S, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 2013; 123: 3243-53. ArticlePubMedPMC
- 48. Ashraf S, Gee HY, Woerner S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest 2013; 123: 5179-89. ArticlePubMedPMC
- 49. Gee HY, Ashraf S, Wan X, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 2014; 94: 884-90. ArticlePubMedPMC
- 50. Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000; 24: 251-6. ArticlePubMedPDF
- 51. Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005; 308: 1801-4. PubMed
- 52. Gigante M, Pontrelli P, Montemurno E, et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant 2009; 24: 1858-64. ArticlePubMed
- 53. Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329: 841-5. PubMedPMC
- 54. Brown EJ, Schlöndorff JS, Becker DJ, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 2010; 42: 72-6. ArticlePubMedPDF
- 55. Mele C, Iatropoulos P, Donadelli R, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 2011; 365: 295-306. PubMedPMC
- 56. Barua M, Stellacci E, Stella L, et al. Mutations in PAX2 associate with adult-onset FSGS. J Am Soc Nephrol 2014; 25: 1942-53. ArticlePubMedPMC
- 57. Gbadegesin RA, Hall G, Adeyemo A, et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 2014; 25: 1991-2002. ArticlePubMedPMC
- 58. Ebarasi L, Ashraf S, Bierzynska A, et al. Defects of CRB2 cause steroid-resistant nephrotic syndrome. Am J Hum Genet 2015; 96: 153-61. ArticlePubMedPMC
- 59. Wagner N, Wagner KD, Xing Y, Scholz H, Schedl A. The major podocyte protein nephrin is transcriptionally activated by the Wilms’ tumor suppressor WT1. J Am Soc Nephrol 2004; 15: 3044-51. ArticlePubMed
- 60. Hinkes BG, Mucha B, Vlangos CN, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007; 119: e907-19. ArticlePubMedPDF
- 61. Machuca E, Benoit G, Nevo F, et al. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol 2010; 21: 1209-17. ArticlePubMedPMC
- 62. Lee JH, Han KH, Lee H, et al. Genetic basis of congenital and infantile nephrotic syndromes. Am J Kidney Dis 2011; 58: 1042-3. ArticlePubMed
- 63. Appel D, Kershaw DB, Smeets B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 2009; 20: 333-43. ArticlePubMedPMC
- 64. Ohse T, Chang AM, Pippin JW, et al. A new function for parietal epithelial cells: a second glomerular barrier. Am J Physiol Renal Physiol 2009; 297: F1566-74. ArticlePubMedPMC
- 65. Sagrinati C, Netti GS, Mazzinghi B, et al. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol 2006; 17: 2443-56. ArticlePubMed
- 66. Ronconi E, Sagrinati C, Angelotti ML, et al. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 2009; 20: 322-32. ArticlePubMedPMC
- 67. Lasagni L, Romagnani P. Glomerular epithelial stem cells: the good, the bad, and the ugly. J Am Soc Nephrol 2010; 21: 1612-9. PubMed
- 68. Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol 2009; 20: 2604-15. ArticlePubMedPMC
- 69. Smeets B, Kuppe C, Sicking EM, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 2011; 22: 1262-74. ArticlePubMedPMC
- 70. Smeets B, Stucker F, Wetzels J, et al. Detection of activated parietal epithelial cells on the glomerular tuft distinguishes early focal segmental glomerulosclerosis from minimal change disease. Am J Pathol 2014; 184: 3239-48. ArticlePubMedPMC
- 71. Fatima H, Moeller MJ, Smeets B, et al. Parietal epithelial cell activation marker in early recurrence of FSGS in the transplant. Clin J Am Soc Nephrol 2012; 7: 1852-8. ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- miRNAs involved in the TGFB signaling as possible markers of steroid-resistant nephrotic syndrome in children
Ahmedz Widiasta, Yunia Sribudiani, Husna Nugrahapraja, Dedi Rachmadi
Gene Reports.2025; 39: 102173. CrossRef - Purinergic Receptor Activation Protects Glomerular Microvasculature from Increased Mechanical Stress in Angiotensin II-Induced Hypertension: A Modeling Study
Owen Richfield, Ricardo Cortez, Supaporn Kulthinee, Martha Franco, L. Gabriel Navar
International Journal of Molecular Sciences.2025; 26(5): 1928. CrossRef - Podocyte A20/TNFAIP3 Controls Glomerulonephritis Severity via the Regulation of Inflammatory Responses and Effects on the Cytoskeleton
Paulina Köhler, Andrea Ribeiro, Mohsen Honarpisheh, Ekaterina von Rauchhaupt, Georg Lorenz, Chenyu Li, Lucas Martin, Stefanie Steiger, Maja Lindenmeyer, Christoph Schmaderer, Hans-Joachim Anders, Dana Thomasova, Maciej Lech
Cells.2025; 14(5): 381. CrossRef - Clinical implications of apolipoprotein L1 testing in patients with focal segmental glomerulosclerosis: a review of diagnostic and prognostic implications
Aiman Waheed, Muhammad Hamza Gul, Risha Naeem, Sardar Noman Qayyum, Khizra Batool, Abeeha Shaukat, Nashmiya Khan, Safa Irfan Shah, Aisha Rehman Siddiqui, Asad Ullah Farooq, Eeshah Nasir, Samim Noori
Annals of Medicine & Surgery.2025; 87(3): 1543. CrossRef - Focal Segmental Glomerulosclerosis: Comprehensive Review and Exploration of the Dual Potential of Cyclodextrins in Therapeutic Optimization
Filipa Mascarenhas-Melo, Bruna Martins, Inês Monteiro, Alka Lohani, Karolline Krambeck
International Journal of Molecular Sciences.2025; 26(18): 8760. CrossRef - Focal segmental glomerulosclerosis associated with undescribed mutation in the LMX1B gene
María Adoración Martín Gómez, Mercedes Caba Molina, Miriam León Fradejas, Juana Alonso Titos, Rafael del Pozo Alvarez
European Journal of Medical Genetics.2024; 72: 104980. CrossRef - MicroRNA-155-5p Aggravates Adriamycin-Induced Focal Segmental Glomerulosclerosis through Targeting Nrf2
Guoyong Liu, Liyu He, Xiaomeng Yang, Lingling Tang, Wei Shi, Jian She, Jiali Wei
Nephron.2023; 147(2): 108. CrossRef - The role of HLA antigens in recurrent primary focal segmental glomerulosclerosis
Ibrahim Batal, Pascale Khairallah, Astrid Weins, Nicole K. Andeen, Michael B. Stokes
Frontiers in Immunology.2023;[Epub] CrossRef - IgM and C3 Deposition in Primary Focal Segmental Glomerulosclerosis (FSGS): A Clinical and Histopathological Spectrum
Faizan Amer, Madiha Syed , Aurangzeb Afzal, Mudassar Hussain , Usman Hassan, Shaarif Bashir, Maryam Hameed, Sheeba Ishtiaq
Cureus.2023;[Epub] CrossRef - Identification of key biomarkers of the glomerulus in focal segmental glomerulosclerosis and their relationship with immune cell infiltration based on WGCNA and the LASSO algorithm
Yun Xia Zhang, Juan Lv, Jun Yuan Bai, XiaoWei Pu, En Lai Dai
Renal Failure.2023;[Epub] CrossRef - Rituximab in the treatment of primary FSGS: time for its use in routine clinical practice?
Adam D Morris, Lauren Floyd, Alexander Woywodt, Ajay Dhaygude
Clinical Kidney Journal.2023; 16(8): 1199. CrossRef - High Rate of Mutations of Adhesion Molecules and Extracellular Matrix Glycoproteins in Patients with Adult-Onset Focal and Segmental Glomerulosclerosis
Sara Marcos González, Emilio Rodrigo Calabia, Ignacio Varela, Michal Červienka, Javier Freire Salinas, José Javier Gómez Román
Biomedicines.2023; 11(6): 1764. CrossRef - Glomerular parietal epithelial expression of CD44 in minimal change nephrotic syndrome and primary focal segmental glomerulosclerosis: A clinico-pathological study
ENithin Paul, Suchitha Satish, KiranKrishnamurthy Kelur, ManjunathSanjeev Shetty
Indian Journal of Pathology and Microbiology.2023; 66(3): 526. CrossRef - Calcineurin inhibitors or cyclophosphamide in the treatment of membranous nephropathy superimposed with FSGS lesions: a retrospective study from China
Hong-Guang He, Yi-Yun Huang, Qin-Qing Liang, Qiu-Rong Ye, An-Dong Li, Kun Ye, Qiu-Xia Wu, Yan-Wu You
Renal Failure.2023;[Epub] CrossRef - CD44-negative parietal–epithelial cell staining in minimal change disease: association with clinical features, response to corticosteroids and kidney outcome
Neus Roca, Elias Jatem, Anabel Abo, Maria Santacana, Alejandro Cruz, Álvaro Madrid, Gloria Fraga, Marisa Martin, Jorge Gonzalez, Cristina Martinez, Anna Balius, Alfons Segarra
Clinical Kidney Journal.2022; 15(3): 545. CrossRef - Monogenic focal segmental glomerulosclerosis: A conceptual framework for identification and management of a heterogeneous disease
Meenakshi Sambharia, Prerna Rastogi, Christie P. Thomas
American Journal of Medical Genetics Part C: Seminars in Medical Genetics.2022; 190(3): 377. CrossRef - Small Nucleolar RNAs in Pseudoexfoliation Glaucoma
Karolina Gasińska, Marcin Czop, Ewa Kosior-Jarecka, Dominika Wróbel-Dudzińska, Janusz Kocki, Tomasz Żarnowski
Cells.2022; 11(17): 2738. CrossRef - Collapsing focal segmental glomerulosclerosis in a patient with oral cavity cancer
Sae Byeol Choi, Kyoung Min Kim, Moon Hyang Park, Kyung Pyo Kang
Medicine.2021; 100(18): e25857. CrossRef - MRTF: Basic Biology and Role in Kidney Disease
Maria Zena Miranda, Zsuzsanna Lichner, Katalin Szászi, András Kapus
International Journal of Molecular Sciences.2021; 22(11): 6040. CrossRef - The Unique Difference Between Serum Level of Soluble Urokinase Plasminogen Activator Receptor (suPAR) in Steroid-Resistant Nephrotic Syndrome Children Treated with an Alkylating Agent and Calcineurin Inhibitors
Ahmedz Widiasta, Kurnia Wahyudi, Husna Nugrahapraja, Yunia Sribudiani, Dedi Rachmadi
Journal of Comprehensive Pediatrics.2021;[Epub] CrossRef - Recessive, gain-of-function toxicity in an APOL1 BAC transgenic mouse model mirrors human APOL1 kidney disease
Gizelle M. McCarthy, Angelo Blasio, Olivia G. Donovan, Lena B. Schaller, Althea Bock-Hughes, Jose M. Magraner, Jung Hee Suh, Calum F. Tattersfield, Isaac E. Stillman, Shrijal S. Shah, Zsuzsanna K. Zsengeller, Balajikarthick Subramanian, David J. Friedman,
Disease Models & Mechanisms.2021;[Epub] CrossRef - CLEC14A protects against podocyte injury in mice with adriamycin nephropathy
Zeyu Su, Yujia Li, Hang Lv, Xiaoyang Cui, Min Liu, Ziying Wang, Yan Zhang, Junhui Zhen, Wei Tang, Xiaojie Wang, Fan Yi
The FASEB Journal.2021;[Epub] CrossRef - Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis
Sandra Rayego-Mateos, Sofia Campillo, Raúl R. Rodrigues-Diez, Antonio Tejera-Muñoz, Laura Marquez-Exposito, Roel Goldschmeding, Diego Rodríguez-Puyol, Laura Calleros, Marta Ruiz-Ortega
Clinical Science.2021; 135(16): 1999. CrossRef - The recruitment mechanisms and potential therapeutic targets of podocytes from parietal epithelial cells
Lihua Ni, Cheng Yuan, Xiaoyan Wu
Journal of Translational Medicine.2021;[Epub] CrossRef - miR-150 inhibitor ameliorates adriamycin-induced focal segmental glomerulosclerosis
Huimeng Qi, Jingqi Fu, Junjun Luan, Congcong Jiao, Xiangfei Cui, Xiangyan Cao, Yixiao Zhang, Yanqiu Wang, Jeffrey B. Kopp, Jingbo Pi, Hua Zhou
Biochemical and Biophysical Research Communications.2020; 522(3): 618. CrossRef - From protein uptake to Dent disease: An overview of the CLCN5 gene
Lisa Gianesello, Dorella Del Prete, Monica Ceol, Giovanna Priante, Lorenzo Arcangelo Calò, Franca Anglani
Gene.2020; 747: 144662. CrossRef - Non-ischemic cardiomyopathy with focal segmental glomerulosclerosis
Parminder Kaur, Balraj Singh, Prem Patel, Rahul Vasudev, Upamanyu Rampal, Fayez Shamoon
Journal of Community Hospital Internal Medicine Perspectives.2020; 10(2): 154. CrossRef - Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases
Marloes Sol, Jan A. A. M. Kamps, Jacob van den Born, Marius C. van den Heuvel, Johan van der Vlag, Guido Krenning, Jan-Luuk Hillebrands
Frontiers in Pharmacology.2020;[Epub] CrossRef - A novel heterozygous variant of the COL4A4 gene in a Chinese family with hematuria and proteinuria leads to focal segmental glomerulosclerosis and chronic kidney disease
Liang‐Liang Fan, Lv Liu, Fang‐Mei Luo, Ran Du, Chen‐Yu Wang, Yi Dong, Ji‐Shi Liu
Molecular Genetics & Genomic Medicine.2020;[Epub] CrossRef - Cellular and molecular mechanisms of kidney fibrosis
Sonja Djudjaj, Peter Boor
Molecular Aspects of Medicine.2019; 65: 16. CrossRef - Albumin induces CD44 expression in glomerular parietal epithelial cells by activating extracellular signal‐regulated kinase 1/2 pathway
Xueying Zhao, Xiaoming Chen, Ashmeer Chima, Yuanyuan Zhang, Jasmine George, Alyssa Cobbs, Nerimiah Emmett
Journal of Cellular Physiology.2019; 234(5): 7224. CrossRef - Analysis of the genomic architecture of a complex trait locus in hypertensive rat models links Tmem63c to kidney damage
Angela Schulz, Nicola Victoria Müller, Nina Anne van de Lest, Andreas Eisenreich, Martina Schmidbauer, Andrei Barysenka, Bettina Purfürst, Anje Sporbert, Theodor Lorenzen, Alexander M Meyer, Laura Herlan, Anika Witten, Frank Rühle, Weibin Zhou, Emile de H
eLife.2019;[Epub] CrossRef - Podocyte-Specific Sialylation-Deficient Mice Serve as a Model for Human FSGS
Kristina M. Niculovic, Linda Blume, Henri Wedekind, Elina Kats, Iris Albers, Stephanie Groos, Markus Abeln, Jessica Schmitz, Esther Beuke, Jan H. Bräsen, Anette Melk, Mario Schiffer, Birgit Weinhold, Anja K. Münster-Kühnel
Journal of the American Society of Nephrology.2019; 30(6): 1021. CrossRef - Inhibition of the ERK1/2-mTORC1 axis ameliorates proteinuria and the fibrogenic action of transforming growth factor-β in Adriamycin-induced glomerulosclerosis
Ranjan Das, Soo-Jin Kim, Nhung Thi Nguyen, Hyeong Ju Kwon, Seung-Kuy Cha, Kyu-Sang Park
Kidney International.2019; 96(4): 927. CrossRef - Mechanisms of Scarring in Focal Segmental Glomerulosclerosis
Jianyong Zhong, Jacob B. Whitman, Hai-Chun Yang, Agnes B. Fogo
Journal of Histochemistry & Cytochemistry.2019; 67(9): 623. CrossRef - A bigenic mouse model of FSGS reveals perturbed pathways in podocytes, mesangial cells and endothelial cells
Andrew S. Potter, Keri Drake, Eric W. Brunskill, S. Steven Potter, Peter Hohenstein
PLOS ONE.2019; 14(8): e0216261. CrossRef - Chronic kidney disease: a review of proteomic and metabolomic approaches to membranous glomerulonephritis, focal segmental glomerulosclerosis, and IgA nephropathy biomarkers
Amir Taherkhani, Reyhaneh Farrokhi Yekta, Maede Mohseni, Massoud Saidijam, Afsaneh Arefi Oskouie
Proteome Science.2019;[Epub] CrossRef - Sphingolipid signaling in renal fibrosis
Andrea Huwiler, Josef Pfeilschifter
Matrix Biology.2018; 68-69: 230. CrossRef - A novel assay provides sensitive measurement of physiologically relevant changes in albumin permeability in isolated human and rodent glomeruli
Sara Desideri, Karen L. Onions, Yan Qiu, Raina D. Ramnath, Matthew J. Butler, Christopher R. Neal, Matthew L.R. King, Andrew E. Salmon, Moin A. Saleem, Gavin I. Welsh, C. Charles Michel, Simon C. Satchell, Andrew H.J. Salmon, Rebecca R. Foster
Kidney International.2018; 93(5): 1086. CrossRef - Recent advances of animal model of focal segmental glomerulosclerosis
Jae Won Yang, Anne Katrin Dettmar, Andreas Kronbichler, Heon Yung Gee, Moin Saleem, Seong Heon Kim, Jae Il Shin
Clinical and Experimental Nephrology.2018; 22(4): 752. CrossRef - Urinary podocyte-associated molecules and albuminuria in hypertension
Javier Perez-Hernandez, Maria D. Olivares, Elena Solaz, Fernando Martinez, Sergio Martínez-Hervas, Gernot Pichler, Felipe J. Chaves, Josep Redon, Raquel Cortes
Journal of Hypertension.2018; 36(8): 1712. CrossRef - Segmental Sclerosis and Extracapillary Hypercellularity Predict Diabetic ESRD
Amy K. Mottl, Adil Gasim, Fernanda Payan Schober, Yichun Hu, Askia K. Dunnon, Susan L. Hogan, J. Charles Jennette
Journal of the American Society of Nephrology.2018; 29(2): 694. CrossRef - Chlormethine Hydrochloride is Not Inferior to Tacrolimus in Treating Steroid-Resistant Nephrotic Syndrome
Yuan Yang, Li Zhao, Li Xiao, Yumei Liang, Chang Wang, Xiao Fu, Xuejing Zhu, Shuguang Yuan, Jianling Zhu, Xiaoping Zhu, Yinghong Liu, Jun Li, Jian Luo, Fuyou Liu, Lin Sun
Kidney and Blood Pressure Research.2018; 43(1): 68. CrossRef - Multiple Myeloma in a Patient with Focal Segmental Glomerulosclerosis: A Case Report
Ashraf O. Oweis, Sameeha A. Al Shelleh, Najla Aldaoud, Osama Mohammed Alshari, Mousa A. Al-Abbadi
American Journal of Case Reports.2018; 19: 946. CrossRef - Can podocytes be regenerated in adults?
Stuart J. Shankland, Benjamin S. Freedman, Jeffrey W. Pippin
Current Opinion in Nephrology and Hypertension.2017; 26(3): 154. CrossRef - Plasma exchange in kidney transplantation: Still a valuable option for nephrotic syndrome recurrence
Licia Peruzzi, Roberto Albiani, Karol Giancaspero
Transfusion and Apheresis Science.2017; 56(4): 525. CrossRef - Is CD44 in glomerular parietal epithelial cells a pathological marker of renal function deterioration in primary focal segmental glomerulosclerosis?
Brunna Pinto Froes, Stanley de Almeida Araújo, Eduardo Alves Bambirra, Eduardo Araújo Oliveira, Ana Cristina Simões e Silva, Sérgio Veloso Brant Pinheiro
Pediatric Nephrology.2017; 32(11): 2165. CrossRef - Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy
Hani Y. Suleiman, Robyn Roth, Sanjay Jain, John E. Heuser, Andrey S. Shaw, Jeffrey H. Miner
JCI Insight.2017;[Epub] CrossRef - Resveratrol Attenuates Adriamycin-Induced Focal Segmental Glomerulosclerosis through C3aR/C5aR- Sphingosine Kinase 1 Pathway
Guoyong Liu, Qiang Wang, Yan Shi, Xiaofei Peng, Hong Liu, Youming Peng, Liyu He
Pharmacology.2017; 100(5-6): 253. CrossRef - The Multifaceted Role of the Lysosomal Protease Cathepsins in Kidney Disease
Pasquale Cocchiaro, Valeria De Pasquale, Rossella Della Morte, Simona Tafuri, Luigi Avallone, Anne Pizard, Anna Moles, Luigi Michele Pavone
Frontiers in Cell and Developmental Biology.2017;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
Pathogenesis of Focal Segmental Glomerulosclerosis

Fig. 1. Morphologic variants of focal segmental glomerulosclerosis. (A) Capillary collapse and podocyte hyperplasia are characteristic features of the collapsing variant. (B) In the tip variant, segmental lesion involves the glomerular tuft next to the tubular pole. (C) The perihilar variant is diagnosed when sclerosis or hyalinosis are present in perihilar lesion in more than half of the sclerotic glomeruli.
Fig. 1.
Pathogenesis of Focal Segmental Glomerulosclerosis
| Related protein or gene description | |
|---|---|
| NPHS1 | Nephrin |
| NPHS2 | Podocin |
| PLCE1 (NPHS3) | Phospholipase Cε1 |
| WT1 (NPHS4) | Wilms tumor 1 |
| LAMB2 (NPHS5) | Laminin β2 |
| PTPRO (NPHS6) | Protein tyrosine phosphatase receptor type O |
| ARHGDIA (NPHS8) | Rho GDP dissociation inhibitor α |
| ADCK4 (NPHS9) | aarF domain containing kinase 4 |
| EMP2 (NPHS10) | Epithelial membrane protein 2 |
| ACTN4 (FSGS1) | α-Actin-4 |
| TRPC6 (FSGS2) | Transient receptor potential cation channel 6 |
| CD2AP (FSGS3) | CD2-associated protein |
| APOL1 (FSGS4) | Apolipoprotein L1 |
| INF2 (FSGS5) | Inverted formin |
| MYO1E (FSGS6) | Myosin 1E |
| PAX2 (FSGS7) | Paired box gene 2 |
| ANLN (FSGS8) | Anillin |
| CRB2 (FSGS9) | Crumbs homolog 2 |
Table 1. Genes related to FSGS or nephrotic syndrome
FSGS, focal segmental glomerulosclerosis.

