 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 49(6); 2015 > Article
-
Review
Neuroendocrine Tumors of the Female Reproductive Tract: A Literature Review - Yi Kyeong Chun
-
Journal of Pathology and Translational Medicine 2015;49(6):450-461.
DOI: https://doi.org/10.4132/jptm.2015.09.20
Published online: October 13, 2015
Department of Pathology, Cheil General Hospital and Women’s Healthcare Center, Dankook University College of Medicine, Seoul, Korea
- Corresponding Author: Yi Kyeong Chun, MD Department of Pathology, Cheil General Hospital and Women’s Healthcare Center, Dankook University College of Medicine, 17 Seoae-ro 1-gil, Jung-gu, Seoul 04619, Korea Tel: +82-2-2000-7663 Fax: +82-2-2000-7779 E-mail: ykcmd@naver.com
• Received: September 2, 2015 • Revised: September 17, 2015 • Accepted: September 19, 2015
© 2015 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Neuroendocrine tumors of the female reproductive tract are a heterogeneous group of neoplasms that display various histologic findings and biologic behaviors. In this review, the classification and clinicopathologic characteristics of neuroendocrine tumors of the female reproductive tract are described. Differential diagnoses are discussed, especially for non-neuroendocrine tumors showing high-grade nuclei with neuroendocrine differentiation. This review also discusses recent advances in our pathogenetic understanding of these disorders.
- Classification of cervical NETs
- Cervical NETs have been described using various terminologies without strict diagnostic criteria, such as carcinoid tumor, argyrophil cell carcinoma, apudoma, poorly differentiated small cell carcinoid, small cell tumor with neuroepithelial features, neuroendocrine carcinoid tumor, endocrine carcinoma intermediate cell type, small cell undifferentiated carcinoma, oat cell carcinoma, small cell carcinoma, SCNEC, NEC, neuroendocrine features in poorly differentiated and undifferentiated carcinoma, large cell carcinoma, and LCNEC [2-5]. These varied terminologies have led to poor recognition of the incidence, clinicopathologic features, and biologic behavior of cervical NETs.
- The present four-category classification of cervical NETs composed of TC, AC, LCNEC, and SCNEC was established in 1997 by the College of American Pathologists and the National Cancer Institute [4] and has been used as the World Health Organization (WHO) classification scheme since 2003 [6]. SCNEC is by far the most common NET of the cervix, followed by LCNEC. Cervical TCs are extremely rare [6]. Briefly, TCs show trabecular, insular, or sheet-like architectural patterns. The small, round, and uniform tumor cells have a finely granular chromatin pattern and inconspicuous nucleoli. Mitotic activity is exceedingly low. ACs share patterns of growth with TC, but show hypercellularity, cytologic atypia, increased mitotic activity (five to 10 mitotic figures per 10 high power fields [HPFs]), and necrotic foci. LCNECs grow in sheets with organoid, trabecular, or cord-like patterns, often with peripheral palisading and necrosis. These large neoplastic cells have abundant eosinophilic cytoplasm with vesicular high-grade nuclei and prominent nucleoli. This tumor type has more than 10 mitotic figures per 10 HPFs. Immunohistochemical confirmation of neuroendocrine differentiation is required based on neuroendocrine markers, such as chromogranin, synaptophysin, and CD56. SCNECs show small, round or fusiform cells with scant cytoplasm and hyperchromatic nuclei, with finely granular chromatin, and absent or inconspicuous nucleoli. Nuclear molding, numerous mitotic figures, and apoptotic bodies are common. Architectural patterns include nesting, trabeculae, peripheral palisading, rosette formations, and sheet-like growth. Immunohistochemical staining for neuroendocrine markers is not required for diagnosis [4].
- The WHO classification of gastroentero-pancreatic NETs is a three-tiered grading system, primarily based on tumor mitotic activity and Ki-67 labeling index [7]. Ki-67 labeling index is also incorporated in the recent WHO classification of pulmonary NETs [8]. However, the Ki-67 index is not included in the 2014 WHO diagnostic criteria for cervical NETs [6]. Further study is required to validate the correlation between Ki-67 index and clinical outcomes in cervical NETs.
- Carcinoid tumors
- Primary cervical carcinoids are extremely rare, and metastatic carcinoids should be excluded to ensure a diagnosis of primary cervical carcinoid tumor. In 1976, Albores-Saavedra et al. [5] reported 12 cases of ‘carcinoid’ tumor, dividing them into well-differentiated and poorly differentiated types based on microscopic findings. Cancers resembling islet cell tumors or medullary thyroid carcinoma were diagnosed as well-differentiated ‘carcinoid,’ while those similar to oat cell carcinoma of the lung were diagnosed as poorly differentiated ‘carcinoid.’ These authors appear to have used ‘carcinoid’ as a comprehensive term for NET.
- Generally, the prognosis of TC and AC is better than that of LCNEC and SCNEC. The prognosis of cervical TC is uncertain due to confusing usage of diagnostic terminology and limited follow-up data [5]. Cervical AC is regarded as an aggressive tumor, like SCNEC and LCNEC. AC and LCNEC can be differentiated based on mitotic activity, nuclear atypia, and extent of necrosis. Due to the overlapping histologic features of these tumors, differentiating between AC and LCNEC can be problematic [9,10].
- Neuroendocrine carcinomas
- LCNEC and SCNEC comprise about 2% of cervical carcinomas and are highly aggressive, even at early stages [4]. Due to the aggressive clinical behaviors of both SCNEC and LCNEC, the clinical significance of subdividing cervical NECs is uncertain. NECs are regarded by some as a different morphologic expression of the same neoplasm [10,11]. They tend to have early nodal involvement, distant metastasis, and advanced surgical stage at initial diagnosis. The overall prognosis of cervical NECs is worse than that of cervical squamous cell carcinoma or adenocarcinoma of comparable stage [12,13].
- The majority (>90%) of cervical NECs are associated with high-risk human papillomavirus (HPV), with type 18 being the most prevalent. Immunohistochemical staining for p16 is almost always positive in cervical NECs because of this HPV association [14].
- These tumors often coexist with carcinoma in situ, invasive squamous cell carcinoma, or adenocarcinoma. It is important that clinicians do not miss the NEC component, because the prognosis of pure NEC is not significantly different from that of NEC admixed with non-NEC. The trickiest differential diagnosis of SCNEC is a small cell variant of squamous cell carcinoma. The important morphologic features favoring SCNEC are nuclear molding, finely dispersed nuclear chromatin, necrosis, crush artifact, mitosis, and numerous apoptotic bodies [15]. Differential diagnoses of NECs, especially LCNECs, include poorly differentiated squamous carcinoma and adenocarcinoma, basaloid squamous cell carcinoma, undifferentiated carcinoma, embryonal rhabdomyosarcoma, lymphoma, melanoma, and peripheral neuroectodermal tumor [13,15].
- The origin of cervical NETs is not clear. Unlike diffuse idiopathic pulmonary neuroendocrine cell hyperplasia, which is a precursor to pulmonary NETs, there is no defined precursor lesion in the normal endocervix, though isolated neuroendocrine cells are seen in normal endocervical glands [16]. An X-chromosome clonality assay showed monoclonality of both components in a case of mixed LCNEC and mucinous carcinoma. This suggests that LCNEC might have arisen from an invasive mucinous adenocarcinoma [17].
- Immunohistochemical staining
- The diagnosis of SCNEC is based on histologic features. Neuroendocrine markers do not have to be demonstrated if morphologic findings are suitable for this aggressive tumor. The tumor displays 33% to 100% positivity for neuroendocrine markers [4,13,15,18,19]. Unlike SCNEC of the cervix, a definitive diagnosis of LCNEC requires positive staining of at least one neuroendocrine marker [4,15]. Chromogranin, synaptophysin, and CD56 are commonly used neuroendocrine markers. However, CD56 is considered a less specific marker of neuroendocrine differentiation compared to chromogranin and synaptophysin [11,20]. This staining should be carefully interpreted, as CD56 can be present in non-NECs, such as squamous cell carcinomas and adenocarcinomas [20].
- Neuroendocrine differentiation is common in cervical non-NECs such as adenocarcinoma, adenosquamous carcinoma, and squamous cell carcinoma. Positive staining for chromogranin and synaptophysin has been reported in 14 (20.9%) and 5 (9%) cervical non-NEC cases, respectively [21,22]. Without morphologic features of neuroendocrine differentiation, these cases should not be diagnosed as LCNEC. Controversial results have been reported regarding the clinical significance of neuroendocrine differentiation in otherwise typical carcinomas. Savargaonkar et al. [21] found that chromogranin expression does not influence the clinical behavior of cervical non-NECs. On the contrary, Chavez-Blanco et al. [22] reported that synaptophysin expression seems to be correlated with a poor outcome in cervical non-NECs. Thyroid transcription factor-1 (TTF-1) is commonly positive (33%–84%) in cervical NECs and might be a useful marker of these tumors, though it cannot distinguish these tumors from primary pulmonary tumors [23,24]. In conjunction with neuroendocrine markers, p63 is useful in distinguishing between squamous cell carcinoma and small or large cell NECs [24]. Focal or diffuse p63 positivity is seen in 43% of cervical NECs, illustrating that this marker is not specific for squamous differentiation [24,25]. p53 protein is expressed in 43% of cervical NECs [26].
NEUROENDOCRINE TUMORS OF THE UTERINE CERVIX
- NETs of the endometrium include TC, SCNEC, and LCNEC. Only three cases of primary endometrial TCs have been reported in the English literature [27-29]. One International Federation of Gynecology and Obstetrics (FIGO) stage 1 case showed vaginal recurrence approximately six and a half years after the initial presentation [29]. To the best of our knowledge, endometrial AC has not been described in the English literature.
- Neuroendocrine carcinomas
- SCNEC and LCNEC of the endometrium are very uncommon, representing only 0.8% of endometrial cancers [30]. These are highly aggressive tumors with a propensity for systemic spread and poor prognosis. The tumors usually form bulky, intraluminal masses with deep myometrial invasion.
- SCNEC of the endometrium requires morphologically prototypic features of small cell carcinoma, unequivocal evidence of endometrial origin, and immunohistochemical staining of at least one neuroendocrine marker, according to the diagnostic criteria proposed by van Hoeven et al. [31]. However, as with other sites, a small number of SCNEC cases show distinctive histologic features of small cell carcinoma without any immunohistochemical evidence of neuroendocrine differentiation [15].
- Endometrial NECs are often combined with other epithelial neoplasms. Endometrioid carcinoma is the most common non-NEC component, and 50% to 80% of NEC cases are admixed with FIGO grade 1 or 2 endometrioid carcinoma. Mixed NEC and conventional endometrial carcinoma can be misinterpreted as FIGO grade 3 endometrioid carcinoma and dedifferentiated carcinoma [30,32,33]. The frequent association of NEC with low-grade endometrioid carcinoma suggests that some endometrial NECs may arise from neuroendocrine cells in endometrioid carcinomas. Interestingly, scattered neuroendocrine cells are reported in the normal endometrial gland and in endometrial carcinoma [34]. These tumors can also form from pluripotent stem cells of the epithelium, which have a capacity for both neuroendocrine and endometrioid glandular differentiation. Lastly, a collision tumor could arise from separate epithelial and NETs [35].
- NEC might be a part of the carcinoma component of carcinosarcoma [36,37]. Uncommon endometrial tumors composed of papillary serous carcinoma and small cell carcinoma have been reported [35,38].
- Differential diagnoses of NEC
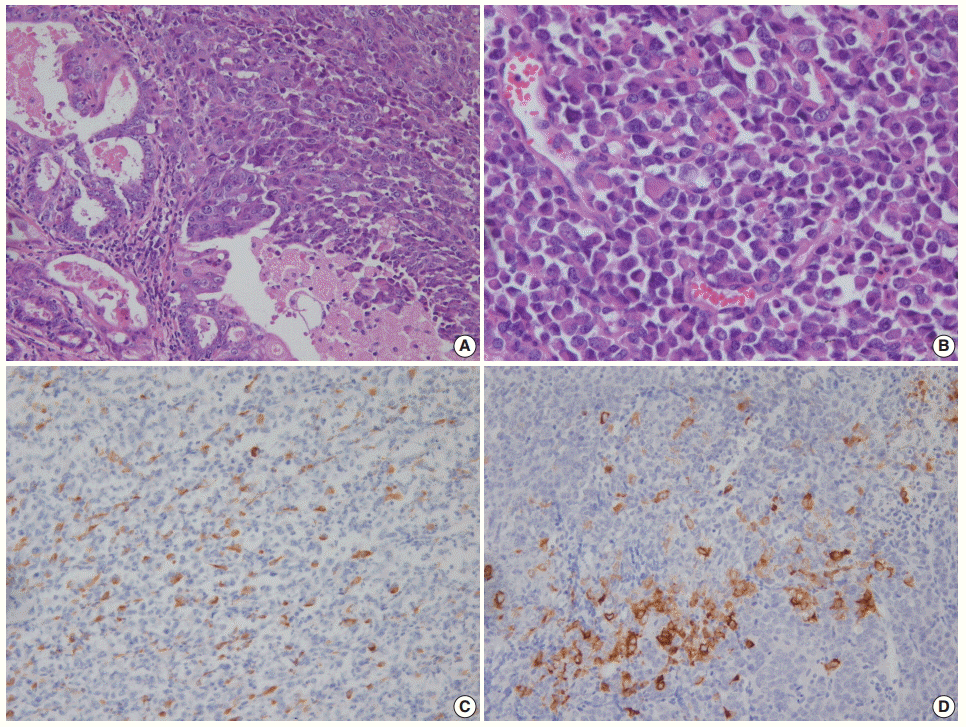
- NECs of the endometrium should be differentiated from various tumors showing high-grade nuclear features with a predominantly solid growth pattern. LCNEC is much more difficult to diagnose than SCNEC, as the tumor might not show the characteristic morphologic features of neuroendocrine differentiation, such as hyperchromatic nuclei, salt and pepper chromatin, and nuclear molding. To establish a diagnosis of endometrial LCNEC, neuroendocrine patterns (nesting, trabeculae, rosettes, and palisading) should be present in at least part of the tumor, along with expression of one or more of the neuroendocrine markers (Fig. 1).
- Undifferentiated endometrial carcinoma is defined as a malignant epithelial neoplasm with no evidence of differentiation. When undifferentiated carcinoma is associated with a well to moderately differentiated endometrioid carcinoma, it should be diagnosed as a dedifferentiated carcinoma. Dedifferentiated carcinoma does not appear to confer better clinical outcomes than undifferentiated carcinoma, despite the presence of a better differentiated non-NEC component. As NEC is often accompanied by low-grade endometrioid carcinoma, it can be misinterpreted as dedifferentiated carcinoma. In addition, dedifferentiated carcinoma is frequently misdiagnosed as NEC, FIGO grade 2 or 3 endometrioid carcinoma, carcinosarcoma, high-grade endometrial stromal sarcoma, lymphoma, granulosa cell tumor, or epithelioid sarcoma [39].
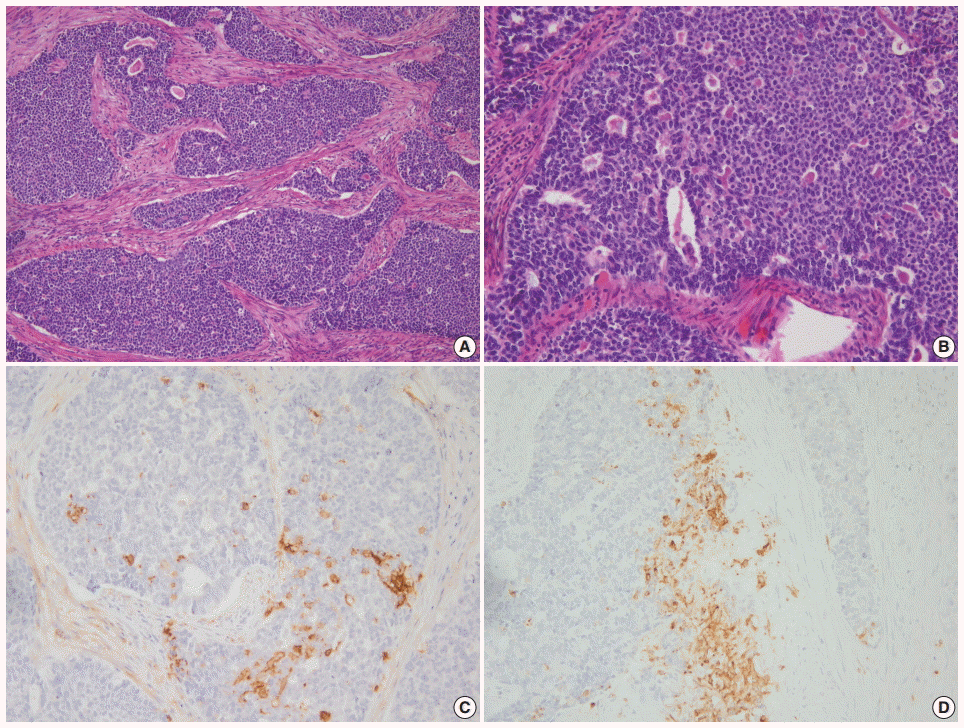
- Morphologically undifferentiated carcinoma is composed of small to intermediate-sized, dyscohesive cells growing in a patternless fashion without gland formation. Most cases have necrosis and more than 25 mitotic figures per 10 HPFs. Immunohistochemical staining for cytokeratin and epithelial membrane antigen (EMA) shows focal positivity, usually in less than 10% of tumor cells. Dedifferentiated carcinoma has different cytologic features in the undifferentiated carcinoma and endometrioid carcinoma components (Fig. 2). On the contrary, poorly differentiated endometrioid carcinoma shows similar tumor cells in the solid and glandular areas (Fig. 3). The solid area often resembles poorly differentiated non-keratinizing squamous cell carcinoma and tends to have a cohesive appearance and diffuse positivity for cytokeratin and EMA [40]. Poorly differentiated endometrioid carcinoma should be distinguished from NEC and undifferentiated/dedifferentiated carcinoma [41]. This distinction has important clinical implications, as endometrioid carcinoma confers a much better prognosis than NEC and undifferentiated/ dedifferentiated carcinoma [42].
- Serous carcinoma with a solid growth pattern and massive necrosis should be differentiated from LCNEC, dedifferentiated carcinoma, and poorly differentiated endometrioid carcinoma. The serous component is negative for neuroendocrine markers, but diffuse expressions of p16 and p53 have been reported in both serous carcinoma and NEC [43]. Along with neuroendocrine makers, it is important to find diagnostic foci of classical serous carcinoma even when the tumor is predominantly solid [38].
- Neuroendocrine expression in non-NECs
- Non-NECs of the endometrium can express neuroendocrine markers but lack typical neuroendocrine histomorphology. Expression of neuroendocrine markers is reported in 62.5% of FIGO grade 3 endometrioid carcinomas, which are more frequently associated with deep myometrial invasion, metastasis to distant organs, and decreased survival than tumors without neuroendocrine expression. A high-grade tumor with diffuse, strong neuroendocrine positivity should be classified as a NEC rather than a poorly differentiated endometrioid carcinoma [44].
- Expression of neuroendocrine markers is reported in 30% and 41% of undifferentiated carcinomas, and most cases exhibit focal neuroendocrine expression in less than 10% of the cells [39,45]. There is no reported difference in overall survival with or without neuroendocrine differentiation in undifferentiated carcinomas. Undifferentiated carcinoma is a highly aggressive tumor, regardless of neuroendocrine expression [40,45].
NEUROENDOCRINE TUMORS OF THE ENDOMETRIUM
- Carcinoid tumors
- Ovarian carcinoid tumors are monodermal teratomas occurring in a pure form (15%) or combined with other teratomatous components (85%), such as a dermoid cyst or a struma ovarii. They can also be a component of mucinous and Brenner tumors. Carcinoid tumors of the ovary can be primary or metastatic; these metastases are usually from gastrointestinal tumors. Primary ovarian carcinoids are mostly confined to a unilateral ovary and behave in an indolent fashion, whereas metastatic tumors tend to be aggressive and associated with poor outcome. Therefore, the distinction between ovarian primary and metastatic carcinoids is critical. In addition to a clinical history of carcinoid tumor in an extraovarian site, such as the gastrointestinal tract or lung, metastatic carcinoids more often show bilateral distribution, multinodular growth, extraovarian tumor nodules, lymphovascular invasion, and absence of teratomatous elements. Ovarian carcinoids can be confused with other primary ovarian tumors, particularly Brenner tumors, granulosa cell tumors, and Sertoli or Sertoli-Leydig cell tumors [15,46-51].
- Primary carcinoid tumors of the ovary are divided into insular, trabecular, strumal, and mucinous carcinoids. Mixed forms include carcinoid tumors that contain two or more of the aforementioned categories and those that are mixed other types of primary ovarian tumors. Briefly, insular carcinoid, considered to be of midgut derivation, is the most common type of primary ovarian carcinoid tumor. It is composed of small acini and solid nests of round cells with uniform nuclei and abundant eosinophilic cytoplasm. Carcinoid syndrome occurs in about one-third of patients with insular carcinoid, despite the absence of metastasis [48]. Trabecular carcinoid, considered to be of hindgut or foregut derivation, shows wavy ribbons or a trabecular arrangement of cells in a dense fibrous stroma. The tumors cells are one or two layers thick, and the nuclei are perpendicular to the axis of the ribbon or the trabeculae. Strumal carcinoid is characterized by the coexistence of carcinoid and thyroid tissue. Mucinous carcinoid, the least common type of ovarian carcinoid, resembles a goblet cell carcinoid arising in the appendix. It must be distinguished from a Krukenberg tumor and has been subdivided into well differentiated mucinous carcinoid, atypical mucinous carcinoid, carcinoma arising in mucinous carcinoid, and mixed mucinous carcinoid and other carcinoid types [15,46-51].
- Primary ovarian carcinoid tumors confined to the ovary and treated with surgery alone are expected to have an excellent overall outcome [47]. Robboy et al. [48] reported two recurrences in 48 cases of primary insular carcinoid of the ovary and calculated a survival rate of 95% and 88% at 5 and 10 years, respectively. Mucinous carcinoids might have more aggressive behavior than other types of ovarian carcinoids, particularly if associated with atypical features [50]. Some ovarian AC cases have been misdiagnosed as ‘carcinoid’ or ‘strumal carcinoid [52,53].’ Kurabayashi et al. [53] reported a case of stage IA strumal ‘carcinoid’ tumor showing multiple bone and breast metastases 3.5 years postoperatively. Histologic features of the case were consistent with AC. The term ‘AC’ is not included in the past or current WHO classification of ovarian NETs [51,54]. Division into four subcategories (insular, trabecular, strumal, and mucinous) instead of TC and AC has complicated the comparison between follow-up data from ovarian carcinoids and carcinoids from other organs. Further study is clearly necessary to better understand the clinical course of ovarian carcinoids.
- Carcinoid tumors are immunoreactive to neuroendocrine markers, such as chromogranin, synaptophysin, and CD56. Chromogranin and synaptophysin are excellent discriminatory neuroendocrine markers for a carcinoid tumor. In a study of 42 carcinoid tumors, chromogranin, synaptophysin, and CD56 were expressed in 100%, 98%, and 57% of samples, respectively. CD56 was also positive in 48% of Sertoli cell tumors and in 25% of endometrioid carcinomas. CD56 is neither highly sensitive nor specific enough for neuroendocrine lineage and is of limited value in the identification of ovarian carcinoid tumors [55]. Various peptide hormones such as serotonin, gastrin, pancreatic polypeptide, glucagon, vasoactive intestinal peptide, prolactin, and somatostatin can be detected in about 25% of cases [56]. Estrogen receptors and progesterone receptors are usually negative in carcinoid tumors. CDX2, TTF-1, PAX8, and cytokeratins 7 and 20 are used for the discrimination of primary and metastatic carcinoids. As ovarian carcinoid can arise from various teratomatous elements, such as the midgut, hindgut, and respiratory epithelium, the interpretation of immunohistochemical staining should be conducted very prudently [57,58].
- Small cell carcinoma, pulmonary type
- Two types of clinically and histologically distinct small cell carcinoma of the ovary have been described: small cell carcinoma, hypercalcemic type (SCCOHT), and small cell carcinoma, pulmonary type (SCCOPT). Clinical features favoring SCCOPT include older age and the absence of hypercalcemia. Histologically, SCCOPT shows characteristic features of SCNEC, such as finely dispersed chromatin, inconspicuous nucleoli, and nuclear molding, whereas SCCOHT has clumped chromatin, prominent nucleoli, and the presence of larger cells in about 50% of cases. Follicle-like spaces are frequently seen in the hypercalcemic type, but are lacking in the pulmonary type. Ovarian surface epithelial tumors are present in more than 50% of pulmonary type tumors, but are absent in the hypercalcemic type [59].
- SCCOPT is a highly aggressive SCNEC and must be distinguished from metastatic small cell carcinoma from other locations, particularly the lung. Usually, bilateral ovarian involvement is a substantial clue for a metastatic tumor, but is also seen in 45% of SCCOPTs. As this tumor shows variable TTF-1 expression, expression of this marker cannot reliably distinguish SCCOPT from pulmonary small cell carcinoma [60]. These tumors are probably of surface epithelial-stromal origin because they are frequently associated with surface epithelial tumors. In a previous study, eight of 11 SCCOPT cases were associated with surface epithelial tumors [59]. Rare cases arising in an ovarian teratoma have been reported [61,62]. A diagnosis of SCCOPT can be made in the absence of neuroendocrine marker positivity if the morphologic appearance is typical SCNEC. Perinuclear dot-like cytokeratin 20 staining has been reported in this tumor, as in Merkel cell carcinoma and salivary gland small cell carcinoma [63].
- Large cell neuroendocrine carcinoma
- LCNECs of the ovary have also been reported as non-small cell NEC, undifferentiated non-small cell carcinoma, and NEC, non-small cell type [64-67]. Primary ovarian LCNEC is extremely rare and has a worse prognosis than usual ovarian carcinomas, even when the diagnosis is made at an early stage. In most cases, there are concomitant ovarian surface epithelial tumors, such as mucinous borderline tumor or mucinous carcinoma, endometrioid carcinoma, serous carcinoma, unclassified high-grade carcinoma, or teratoma [64-67]. The NEC component varies from 10% to 90% when it is combined with an epithelial tumor or teratoma. The presence of an NEC component in an otherwise usual epithelial tumor should be reported because of the potential negative prognostic impact of NEC histology. Generally speaking, a neuroendocrine component might have a prognostic impact when it reaches a certain proportion of the overall tumor. The percentage of NEC component that is necessary to confer a prognosis worse than that of the accompanying epithelial tumor is not clear [67]. Primary pure LCNEC of the ovary is very rare [68,69].
- LCNECs probably arise from the neuroendocrine cells present in surface epithelial-stromal tumors or germ cell tumors [70]. In a case of mixed LCNEC and mucinous borderline ovarian tumor, clonality analysis using the human androgen receptor gene showed monoclonality in both components, suggesting that the LCNEC might have arisen from the mucinous epithelial tumor [71]. The common coexistence of NEC and epithelial tumors, along with the monoclonality of the two components, implies a common cellular origin of the neuroendocrine and epithelial components [17,72]. A case of LCNEC associated with serous carcinoma revealed a different pattern of microsatellite instability in both components. A dual origin with concomitant transformation of epithelial cells and neuroendocrine cells might be possible in that case [65].
- In one previous study, ovarian LCNECs were misdiagnosed as dysgerminoma, sex cord tumor, or other types of carcinoma in eight of 11 cases [64]. Attention to the histologic features of neuroendocrine differentiation and the use of immunohistochemical stains are necessary to resolve this potential underrecognition [64]. CD56 is known to be a less specific neuroendocrine marker compared to chromogranin and synaptophysin [11,20,55]. However, chromogranin and synaptophysin can be detected in ovarian Sertoli cell tumors, Sertoli-Leydig cell tumors, and endometrioid tumors, which could be a potential pitfall resulting in the misdiagnosis of NETs [55,73].
- LCNEC associated with serous carcinoma has rarely been reported in the ovary, as in the endometrium [65,74]. Immunohistochemical evidence of neuroendocrine differentiation in ovarian serous carcinoma is more frequent than morphological evidence. Taube et al. [75] reported synaptophysin and chromogranin expression in 6.7% and 20.7% of high-grade ovarian serous carcinomas, respectively, and found that patients with synaptophysin expression in more than 20% of tumor cells had a significantly shorter survival time than those with 0% to 20% positive cells.
- Small cell carcinoma, hypercalcemic type
- SCCOHT of the ovary is a highly aggressive neoplasm affecting young females and is associated with paraneoplastic hypercalcemia in two-thirds of cases. Microscopic findings show a sheet-like arrangement of cells punctured by follicle-like spaces. The tumor cells are typically small and round with hyperchromatic nuclei and brisk mitotic activity. A large cell component with moderate to abundant eosinophilic cytoplasm is seen in about 50% of cases. Tumors composed exclusively of large cells are designated the ‘large cell variant’ of SCCOHT. The large cells can have a rhabdoid appearance. Differential diagnoses include juvenile and adult granulosa cell tumors, high-grade serous carcinoma, desmoplastic small round cell tumor, dysgerminoma, Ewing sarcoma, primitive neuroectodermal tumor, neuroblastoma, round cell sarcoma, high-grade endometrial stromal sarcoma, undifferentiated carcinoma, lymphoma, melanoma, and SCNEC [76].
- Ovarian SCCOHT is often misunderstood or confused with a subtype of NEC due to the usage of the term ‘small cell carcinoma’. This tumor is included in a miscellaneous category in the 2014 WHO classification of female reproductive organs [77]. Recently, somatic and germline SMARCA4 mutations accompanied by the loss of BRG1 protein expression in immunohistochemistry have been described in SCCOHTs [78-81]. To date, the diagnosis of SCCOHT has been made on the basis of microscopic findings without any specific immunohistochemical markers. The loss of BRG1 protein expression is confirmed to be a useful marker for diagnosing SCCOHT, although the interpretation should be conducted carefully due to the possible heterogeneity and variable intensity of this immunostaining [82]. BRG1 and INI-1 are members of the SWI/SNF complex and are involved in chromatin remodeling. The alternative expression of INI-1 and BRG1 is regarded as a molecular hallmark of malignant rhabdoid tumor. The histological resemblance between SCCOHT and malignant rhabdoid tumor became a trigger to evaluate INI-1 and BRG1 immunostaining in SCCOHT. In addition to the lack of BRG1 immunoreactivity, SCCOHT cases also showed retained INI-1 expression [78,82,83]. Currently, SCCOHT is considered to be an ovarian malignant rhabdoid tumor [78,82,83].
NEUROENDOCRINE TUMORS OF THE OVARY
- The four-category scheme including TC, AC, LCNEC, and SCNEC is still applied to NETs of the female reproductive tract. Ki-67 labeling index is not included in the diagnostic criteria of the 2014 WHO classification of cervical NETs [6].
- The prevalence and biologic behavior of NETs vary along the female reproductive tract. Carcinoid tumors are extremely rare in the cervix and the endometrium, and their clinical behavior is uncertain due to the scarcity of follow-up data. However, in the ovary, carcinoid tumors are the most common NET. Division into four subcategories (insular, trabecular, strumal, and mucinous) instead of TC and AC has obscured the comparison of follow up data from ovarian carcinoids with that of carcinoids in other organs.
- Both small cell and large cell NECs show highly aggressive clinical behavior, regardless of the site of origin. The uterine cervix is the most common site for NECs, especially SCNECs in the female reproductive tract. Since endometrial NEC is often accompanied by low-grade endometrioid carcinoma, it can be misdiagnosed as FIGO grade 3 endometrioid carcinoma or dedifferentiated carcinoma [30,33]. As NECs are rare and tumors with neuroendocrine differentiation are infrequently found in the endometrium or ovary, various tumors are included in the differential diagnoses. Attention to the histologic features of neuroendocrine differentiation and the immunohistochemical staining of neuroendocrine markers is necessary to reach a correct diagnosis. CD56 is known to be a less specific neuroendocrine marker compared to chromogranin and synaptophysin [11,20,55]. The common coexistence of NEC and epithelial tumors along with the monoclonality of the two components implies a common cellular origin of the neuroendocrine and epithelial components [17,72].
- Ovarian SCCOPT is a highly aggressive SCNEC and must be distinguished from metastatic small cell carcinoma from other locations. Currently, SCCOHT is considered to be an ovarian malignant rhabdoid tumor, as inactivation of SMARCA4 accompanied by the loss of BRG1 protein and the retention of INI- 1 in immunohistochemistry has been described in this aggressive tumor [78,82,83].
CONCLUSION
Acknowledgments
Fig. 1. (A) Endometrial large cell neuroendocrine carcinoma (LCNEC) admixed with grade 1 endometrioid carcinoma. (B) Large vesicular nuclei and prominent nucleoli in LCNEC. (C) CD56 immunostaining: positive in endometrioid carcinoma and negative in LCNEC. (D) Diffuse synaptophysin expression in LCNEC.
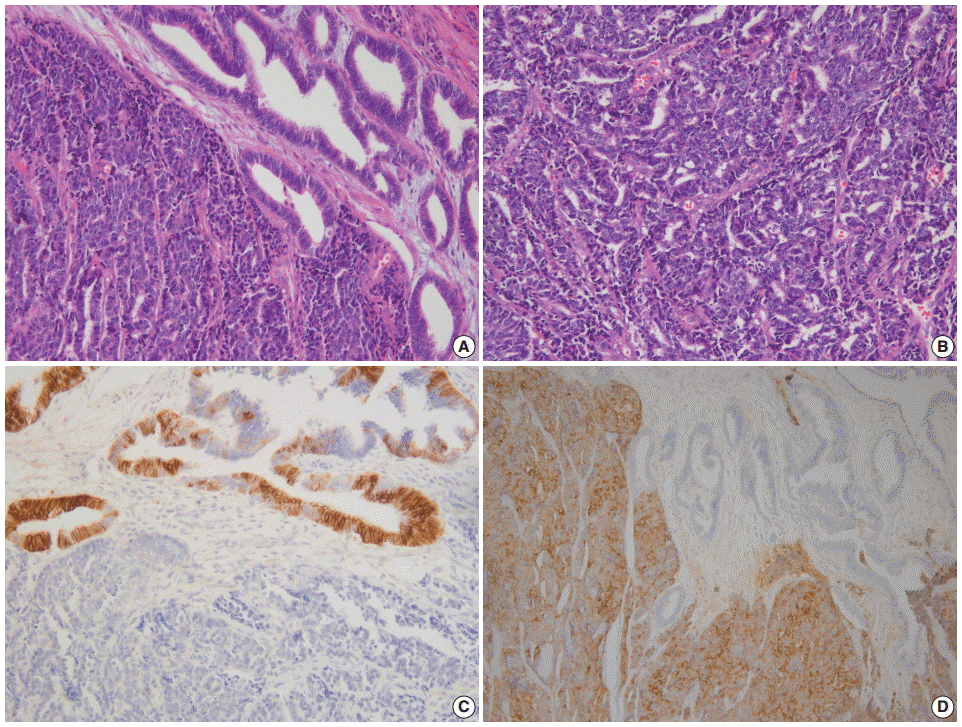
Fig. 2. (A) Dedifferentiated carcinoma composed of undifferentiated carcinoma and grade 1 endometrioid carcinoma. (B) Dyscohesive tumor cells growing in a patternless fashion without gland formation. Focal positivity of cytokeratin (C) and synaptophysin (D) immunostaining.
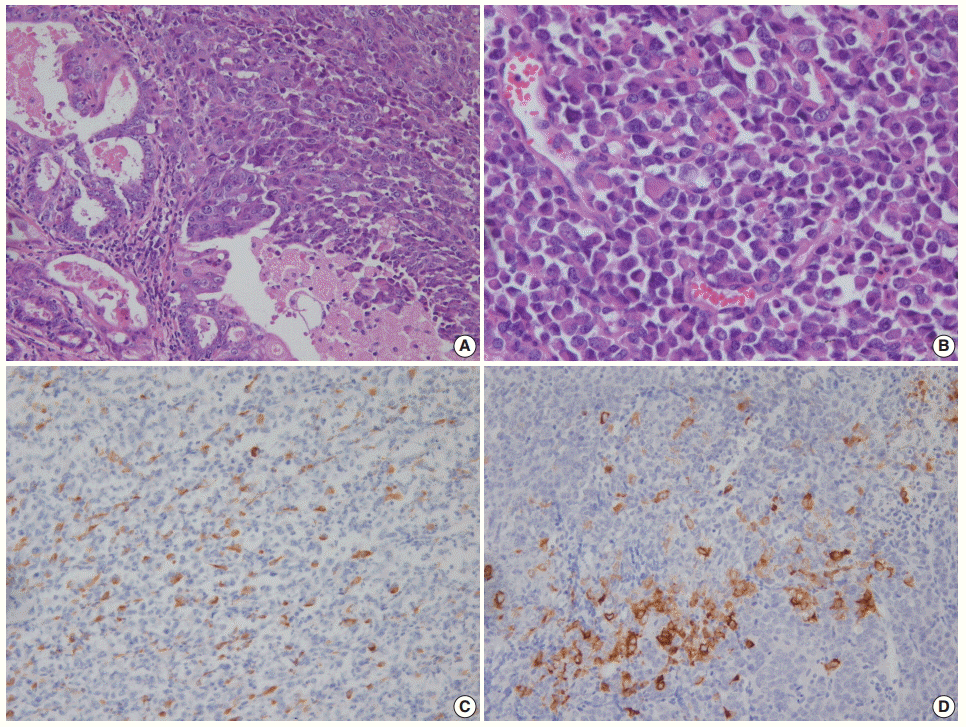
Fig. 3. (A) Grade 3 endometrioid carcinoma mimicking neuroendocrine carcinoma at low magnification. (B) At high magnification, grade 3 endometrioid carcinoma shows similar tumor cells in the solid and glandular areas. Focal positivity for neuroendocrine markers of CD56 (C) and synaptophysin (D).
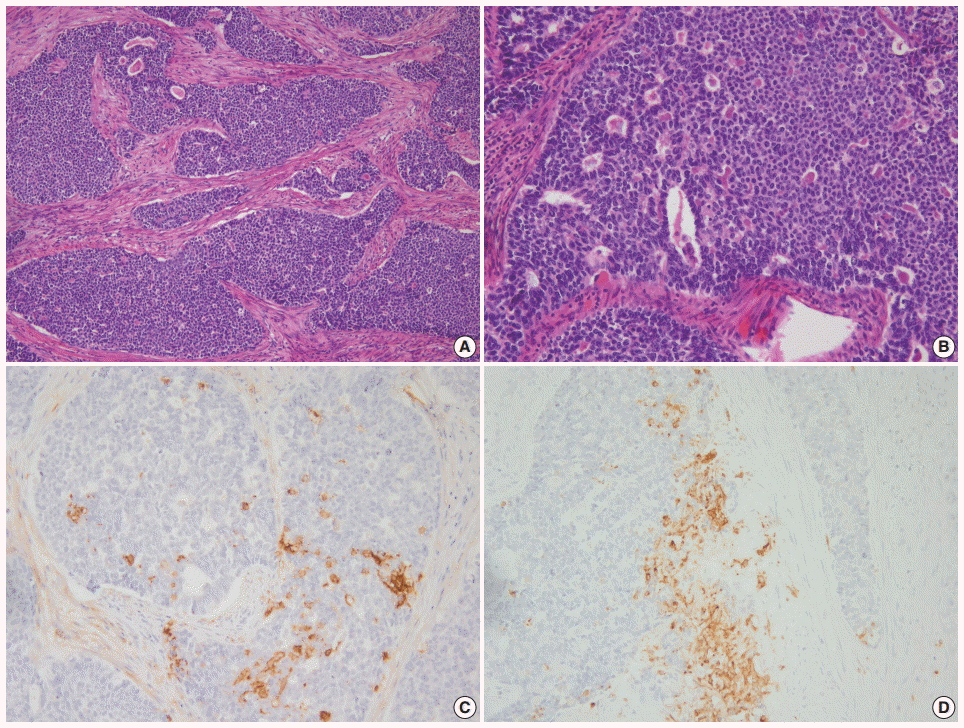
- 1. Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma: an ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol 1991; 15: 529-53. PubMed
- 2. Matsuyama M, Inoue T, Ariyoshi Y, et al. Argyrophil cell carcinoma of the uterine cervix with ectopic production of ACTH, beta-MSH, serotonin, histamine, and amylase. Cancer 1979; 44: 1813-23. ArticlePubMed
- 3. Johannessen JV, Capella C, Solcia E, Davy M, Sobrinho-Simões M. Endocrine cell carcinoma of the uterine cervix. Diagn Gynecol Obstet 1980; 2: 127-34. PubMed
- 4. Albores-Saavedra J, Gersell D, Gilks CB, et al. Terminology of endocrine tumors of the uterine cervix: results of a workshop sponsored by the College of American Pathologists and the National Cancer Institute. Arch Pathol Lab Med 1997; 121: 34-9. PubMed
- 5. Albores-Saavedra J, Larraza O, Poucell S, Rodríguez Martínez HA. Carcinoid of the uterine cervix: additional observations on a new tumor entity. Cancer 1976; 38: 2328-42. ArticlePubMed
- 6. Colgan TJ, Kim I, Hirschowitz L, McCluggage WG. Neuroendocrine tumours. In: Kurman RJ, Carcangiu ML, Herrington CS, Young RH, eds. WHO classification of tumours of female reproductive organs. 4th ed. Lyon: IARC Press, 2014; 196-8.
- 7. Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO classification tumours of the digestive system. 4th ed. Lyon: IARC Press, 2010; 13-4.
- 8. Brambilla E, Gazdar A, Powell CA, et al. Neuroendocrine tumours small cell carcinoma. In: Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG, eds. WHO classification tumours of the lung, pleura, thymus and heart. 4th ed. Lyon: IARC Press, 2015; 63-8.
- 9. Yasuoka T, Hashimoto H, Hamada K, Fujioka T, Nawa A. Atypical carcinoid of the uterine cervix with aggressive clinical behavior: a case report. Gynecol Oncol Case Rep 2014; 7: 4-6. ArticlePubMedPMC
- 10. Witkiewicz AK, Wright TC, Ferenczy A, Ronnett BM, Kurman RJ. Carcinoma and other tumors of the cervix. In: Kurman RJ, Ellenson LH, Ronnett BM, eds. Blaustein’s pathology of the female genital tract. 6th ed. New York: Springer, 2011; 253-304.
- 11. Albores-Saavedra J, Latif S, Carrick KS, Alvarado-Cabrero I, Fowler MR. CD56 reactivity in small cell carcinoma of the uterine cervix. Int J Gynecol Pathol 2005; 24: 113-7. ArticlePubMed
- 12. Atienza-Amores M, Guerini-Rocco E, Soslow RA, Park KJ, Weigelt B. Small cell carcinoma of the gynecologic tract: a multifaceted spectrum of lesions. Gynecol Oncol 2014; 134: 410-8. ArticlePubMed
- 13. Rekhi B, Patil B, Deodhar KK, et al. Spectrum of neuroendocrine carcinomas of the uterine cervix, including histopathologic features, terminology, immunohistochemical profile, and clinical outcomes in a series of 50 cases from a single institution in India. Ann Diagn Pathol 2013; 17: 1-9. ArticlePubMed
- 14. Masumoto N, Fujii T, Ishikawa M, et al. p16 overexpression and human papillomavirus infection in small cell carcinoma of the uterine cervix. Hum Pathol 2003; 34: 778-83. PubMed
- 15. Rouzbahman M, Clarke B. Neuroendocrine tumors of the gynecologic tract: select topics. Semin Diagn Pathol 2013; 30: 224-33. ArticlePubMed
- 16. Koo CW, Baliff JP, Torigian DA, Litzky LA, Gefter WB, Akers SR. Spectrum of pulmonary neuroendocrine cell proliferation: diffuse idiopathic pulmonary neuroendocrine cell hyperplasia, tumorlet, and carcinoids. AJR Am J Roentgenol 2010; 195: 661-8. ArticlePubMed
- 17. Yasuoka H, Tsujimoto M, Ueda M, et al. Monoclonality of composite large-cell neuroendocrine carcinoma and invasive intestinal-type mucinous adenocarcinoma of the cervix: a case study. Int J Gynecol Pathol 2013; 32: 416-20. PubMed
- 18. Kuji S, Hirashima Y, Nakayama H, et al. Diagnosis, clinicopathologic features, treatment, and prognosis of small cell carcinoma of the uterine cervix: Kansai Clinical Oncology Group/Intergroup study in Japan. Gynecol Oncol 2013; 129: 522-7. ArticlePubMed
- 19. Tsunoda S, Jobo T, Arai M, et al. Small-cell carcinoma of the uterine cervix: a clinicopathologic study of 11 cases. Int J Gynecol Cancer 2005; 15: 295-300. ArticlePubMed
- 20. Li JD, Zhuang Y, Li YF, et al. A clinicopathological aspect of primary small-cell carcinoma of the uterine cervix: a single-centre study of 25 cases. J Clin Pathol 2011; 64: 1102-7. ArticlePubMed
- 21. Savargaonkar PR, Hale RJ, Mutton A, Manning V, Buckley CH. Neuroendocrine differentiation in cervical carcinoma. J Clin Pathol 1996; 49: 139-41. ArticlePubMedPMC
- 22. Chavez-Blanco A, Taja-Chayeb L, Cetina L, et al. Neuroendocrine marker expression in cervical carcinomas of non-small cell type. Int J Gynecol Pathol 2002; 21: 368-74. ArticlePubMed
- 23. Ordóñez NG. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am J Surg Pathol 2000; 24: 1217-23. ArticlePubMed
- 24. Houghton O, McCluggage WG. The expression and diagnostic utility of p63 in the female genital tract. Adv Anat Pathol 2009; 16: 316-21. ArticlePubMed
- 25. McCluggage WG, Kennedy K, Busam KJ. An immunohistochemical study of cervical neuroendocrine carcinomas: neoplasms that are commonly TTF1 positive and which may express CK20 and p63. Am J Surg Pathol 2010; 34: 525-32. ArticlePubMed
- 26. Conner MG, Richter H, Moran CA, Hameed A, Albores-Saavedra J. Small cell carcinoma of the cervix: a clinicopathologic and immunohistochemical study of 23 cases. Ann Diagn Pathol 2002; 6: 345-8. ArticlePubMed
- 27. Chetty R, Clark SP, Bhathal PS. Carcinoid tumour of the uterine corpus. Virchows Arch A Pathol Anat Histopathol 1993; 422: 93-5. ArticlePubMedPDF
- 28. González-Bosquet E, González-Bosquet J, García Jiménez A, Gil A, Xercavins J. Carcinoid tumor of the uterine corpus: a case report. J Reprod Med 1998; 43: 844-6. PubMed
- 29. Hong SR, Kim HS, Shim JU. Primary carcinoid tumor of the uterine corpus: a case report. Korean J Pathol 2004; 38: 109-12.
- 30. Huntsman DG, Clement PB, Gilks CB, Scully RE. Small-cell carcinoma of the endometrium: a clinicopathological study of sixteen cases. Am J Surg Pathol 1994; 18: 364-75. PubMed
- 31. van Hoeven KH, Hudock JA, Woodruff JM, Suhrland MJ. Small cell neuroendocrine carcinoma of the endometrium. Int J Gynecol Pathol 1995; 14: 21-9. ArticlePubMed
- 32. Koo YJ, Kim DY, Kim KR, et al. Small cell neuroendocrine carcinoma of the endometrium: a clinicopathologic study of six cases. Taiwan J Obstet Gynecol 2014; 53: 355-9. ArticlePubMed
- 33. Mulvany NJ, Allen DG. Combined large cell neuroendocrine and endometrioid carcinoma of the endometrium. Int J Gynecol Pathol 2008; 27: 49-57. ArticlePubMed
- 34. Satake T, Matsuyama M. Argyrophil cells in normal endometrial glands. Virchows Arch A Pathol Anat Histopathol 1987; 410: 449-54. ArticlePubMedPDF
- 35. Shaco-Levy R, Manor E, Piura B, Ariel I. An unusual composite endometrial tumor combining papillary serous carcinoma and small cell carcinoma. Am J Surg Pathol 2004; 28: 1103-6. ArticlePubMed
- 36. Manivel C, Wick MR, Sibley RK. Neuroendocrine differentiation in mullerian neoplasms. An immunohistochemical study of a “pure” endometrial small-cell carcinoma and a mixed mullerian tumor containing small-cell carcinoma. Am J Clin Pathol 1986; 86: 438-43. PubMed
- 37. Toptas T, Tasova-Yildirim G, Karaveli S, Simsek T. Malignant mixed Mullerian tumor with small cell neuroendocrine differentiation: a case report and review of the literature. Eur J Gynaecol Oncol 2014; 35: 180-4. PubMed
- 38. Posligua L, Malpica A, Liu J, Brown J, Deavers MT. Combined large cell neuroendocrine carcinoma and papillary serous carcinoma of the endometrium with pagetoid spread. Arch Pathol Lab Med 2008; 132: 1821-4. ArticlePubMedPDF
- 39. Tafe LJ, Garg K, Chew I, Tornos C, Soslow RA. Endometrial and ovarian carcinomas with undifferentiated components: clinically aggressive and frequently underrecognized neoplasms. Mod Pathol 2010; 23: 781-9. ArticlePubMedPDF
- 40. Altrabulsi B, Malpica A, Deavers MT, Bodurka DC, Broaddus R, Silva EG. Undifferentiated carcinoma of the endometrium. Am J Surg Pathol 2005; 29: 1316-21. ArticlePubMed
- 41. Bartosch C, Manuel Lopes J, Oliva E. Endometrial carcinomas: a review emphasizing overlapping and distinctive morphological and immunohistochemical features. Adv Anat Pathol 2011; 18: 415-37. PubMed
- 42. Silva EG, Deavers MT, Bodurka DC, Malpica A. Association of low-grade endometrioid carcinoma of the uterus and ovary with undifferentiated carcinoma: a new type of dedifferentiated carcinoma? Int J Gynecol Pathol 2006; 25: 52-8. ArticlePubMed
- 43. Albores-Saavedra J, Martinez-Benitez B, Luevano E. Small cell carcinomas and large cell neuroendocrine carcinomas of the endometrium and cervix: polypoid tumors and those arising in polyps may have a favorable prognosis. Int J Gynecol Pathol 2008; 27: 333-9. ArticlePubMed
- 44. Tamura T, Jobo T, Watanabe J, Kanai T, Kuramoto H. Neuroendocrine features in poorly differentiated endometrioid adenocarcinomas of the endometrium. Int J Gynecol Cancer 2006; 16: 821-6. ArticlePubMed
- 45. Taraif SH, Deavers MT, Malpica A, Silva EG. The significance of neuroendocrine expression in undifferentiated carcinoma of the endometrium. Int J Gynecol Pathol 2009; 28: 142-7. ArticlePubMed
- 46. Reed NS, Gomez-Garcia E, Gallardo-Rincon D, et al. Gynecologic Cancer InterGroup (GCIG) consensus review for carcinoid tumors of the ovary. Int J Gynecol Cancer 2014; 24(9 Suppl 3): S35-41. ArticlePubMed
- 47. Davis KP, Hartmann LK, Keeney GL, Shapiro H. Primary ovarian carcinoid tumors. Gynecol Oncol 1996; 61: 259-65. ArticlePubMed
- 48. Robboy SJ, Norris HJ, Scully RE. Insular carcinoid primary in the ovary: a clinicopathologic analysis of 48 cases. Cancer 1975; 36: 404-18. ArticlePubMed
- 49. Robboy SJ, Scully RE. Strumal carcinoid of the ovary: an analysis of 50 cases of a distinctive tumor composed of thyroid tissue and carcinoid. Cancer 1980; 46: 2019-34. ArticlePubMed
- 50. Baker PM, Oliva E, Young RH, Talerman A, Scully RE. Ovarian mucinous carcinoids including some with a carcinomatous component: a report of 17 cases. Am J Surg Pathol 2001; 25: 557-68. PubMed
- 51. Prat JC, Cao D, Carinelli SG, Nogales FF, Vang R, Zaloudek CJ. Monodermal teratoma and somatic-type tumors arising from a dermoid cyst. In: Kurman RJ, Carcangiu ML, Herrington CS, Young RH, eds. WHO classification of tumours of female reproductive organs. 4th ed. Lyon: IARC Press, 2014; 63-5.
- 52. Kim HS, Yoon G, Jang HI, Song SY, Kim BG. Primary ovarian carcinoid tumor showing unusual histology and nuclear accumulation of beta-catenin. Int J Clin Exp Pathol 2015; 8: 5749-52. PubMedPMC
- 53. Kurabayashi T, Minamikawa T, Nishijima S, et al. Primary strumal carcinoid tumor of the ovary with multiple bone and breast metastases. J Obstet Gynaecol Res 2010; 36: 567-71. ArticlePubMed
- 54. Nogales F, Talerman A, Kubik-Huch RA, Tavassoli FA, Devouassoux-Shisheboran M. Germ cell tumours. In: Tavassoli FA, Devilee P, Kubik-Huch RA, Tavassoli FA, Devouassoux-Shisheboran M, eds. Pathology and genetics of tumours of the breast and female genital organs. 3rd ed. Lyon: IARC Press, 2003; 163-75.
- 55. Zhao C, Bratthauer GL, Barner R, Vang R. Comparative analysis of alternative and traditional immunohistochemical markers for the distinction of ovarian sertoli cell tumor from endometrioid tumors and carcinoid tumor: a study of 160 cases. Am J Surg Pathol 2007; 31: 255-66. PubMed
- 56. Sporrong B, Falkmer S, Robboy SJ, et al. Neurohormonal peptides in ovarian carcinoids: an immunohistochemical study of 81 primary carcinoids and of intraovarian metastases from six mid-gut carcinoids. Cancer 1982; 49: 68-74. ArticlePubMedPDF
- 57. Rabban JT, Lerwill MF, McCluggage WG, Grenert JP, Zaloudek CJ. Primary ovarian carcinoid tumors may express CDX-2: a potential pitfall in distinction from metastatic intestinal carcinoid tumors involving the ovary. Int J Gynecol Pathol 2009; 28: 41-8. ArticlePubMed
- 58. Desouki MM, Lioyd J, Xu H, Cao D, Barner R, Zhao C. CDX2 may be a useful marker to distinguish primary ovarian carcinoid from gastrointestinal metastatic carcinoids to the ovary. Hum Pathol 2013; 44: 2536-41. ArticlePubMed
- 59. Eichhorn JH, Young RH, Scully RE. Primary ovarian small cell carcinoma of pulmonary type: a clinicopathologic, immunohistologic, and flow cytometric analysis of 11 cases. Am J Surg Pathol 1992; 16: 926-38. PubMed
- 60. Grandjean M, Legrand L, Waterkeyn M, et al. Small cell carcinoma of pulmonary type inside a microinvasive mucinous cystadenocarcinoma of the ovary: a case report. Int J Gynecol Pathol 2007; 26: 426-31. PubMed
- 61. Rubio A, Schuldt M, Chamorro C, Crespo-Lora V, Nogales FF. Ovarian small cell carcinoma of pulmonary type arising in mature cystic teratomas with metastases to the contralateral ovary. Int J Surg Pathol 2015; 23: 388-92. ArticlePubMedPDF
- 62. Ikota H, Kaneko K, Takahashi S, et al. Malignant transformation of ovarian mature cystic teratoma with a predominant pulmonary type small cell carcinoma component. Pathol Int 2012; 62: 276-80. ArticlePubMed
- 63. Rund CR, Fischer EG. Perinuclear dot-like cytokeratin 20 staining in small cell neuroendocrine carcinoma of the ovary (pulmonary-type). Appl Immunohistochem Mol Morphol 2006; 14: 244-8. ArticlePubMed
- 64. Veras E, Deavers MT, Silva EG, Malpica A. Ovarian nonsmall cell neuroendocrine carcinoma: a clinicopathologic and immunohistochemical study of 11 cases. Am J Surg Pathol 2007; 31: 774-82. PubMed
- 65. Choi YD, Lee JS, Choi C, Park CS, Nam JH. Ovarian neuroendocrine carcinoma, non-small cell type, associated with serous carcinoma. Gynecol Oncol 2007; 104: 747-52. ArticlePubMed
- 66. Behnam K, Kabus D, Behnam M. Primary ovarian undifferentiated non-small cell carcinoma, neuroendocrine type. Gynecol Oncol 2004; 92: 372-5. ArticlePubMed
- 67. Eichhorn JH, Lawrence WD, Young RH, Scully RE. Ovarian neuroendocrine carcinomas of non-small-cell type associated with surface epithelial adenocarcinomas: a study of five cases and review of the literature. Int J Gynecol Pathol 1996; 15: 303-14. PubMed
- 68. Dundr P, Fischerová D, Povýsil C, Cibula D. Primary pure large-cell neuroendocrine carcinoma of the ovary. Pathol Res Pract 2008; 204: 133-7. ArticlePubMed
- 69. Shakuntala PN, Uma Devi K, Shobha K, Bafna UD, Geetashree M. Pure large cell neuroendocrine carcinoma of ovary: a rare clinical entity and review of literature. Case Rep Oncol Med 2012; 2012: 120727.ArticlePubMedPMCPDF
- 70. Khurana KK, Tornos C, Silva EG. Ovarian neuroendocrine carcinoma associated with a mucinous neoplasm. Arch Pathol Lab Med 1994; 118: 1032-4. PubMed
- 71. Yasuoka H, Tsujimoto M, Fujita S, et al. Monoclonality of composite large cell neuroendocrine carcinoma and mucinous epithelial tumor of the ovary: a case study. Int J Gynecol Pathol 2009; 28: 55-8. ArticlePubMed
- 72. Voutsadakis IA. Large cell neuroendocrine carcinoma of the ovary: a pathologic entity in search of clinical identity. World J Clin Oncol 2014; 5: 36-8. ArticlePubMedPMC
- 73. Ki EY, Park JS, Lee KH, Bae SN, Hur SY. Large cell neuroendocrine carcinoma of the ovary: a case report and a brief review of the literature. World J Surg Oncol 2014; 12: 314.ArticlePubMedPMCPDF
- 74. Draganova-Tacheva RA, Khurana JS, Huang Y, Hernandez E, Zhang X. Large cell neuroendocrine carcinoma of the ovary associated with serous carcinoma with mucin production: a case report and literature review. Int J Clin Exp Pathol 2009; 2: 304-9. PubMedPMC
- 75. Taube ET, Denkert C, Pietzner K, Dietel M, Sehouli J, Darb-Esfahani S. Prognostic impact of neuroendocrine differentiation in high-grade serous ovarian carcinoma. Virchows Arch 2015; 466: 333-42. ArticlePubMedPDF
- 76. Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type: a clinicopathological analysis of 150 cases. Am J Surg Pathol 1994; 18: 1102-16. PubMed
- 77. McCluggage WG, Daya D, Ip P, Malpica A, Oliva E, Young RH. Miscellaneous tumours. In: Kurman RJ, Carcangiu ML, Herrington CS, Young RH, eds. WHO classification of tumours of female reproductive organs. 4th ed. Lyon: IARC Press, 2014; 69-73.
- 78. Kupryjan´czyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J, et al. Ovarian small cell carcinoma of hypercalcemic type: evidence of germline origin and SMARCA4 gene inactivation. a pilot study. Pol J Pathol 2013; 64: 238-46. PubMed
- 79. Jelinic P, Mueller JJ, Olvera N, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014; 46: 424-6. ArticlePubMedPMCPDF
- 80. Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 2014; 46: 427-9. ArticlePubMedPMCPDF
- 81. Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014; 46: 438-43. PubMed
- 82. Karanian-Philippe M, Velasco V, Longy M, et al. SMARCA4 (BRG1) loss of expression is a useful marker for the diagnosis of ovarian small cell carcinoma of the hypercalcemic type (ovarian rhabdoid tumor): a comprehensive analysis of 116 rare gynecologic tumors, 9 soft tissue tumors, and 9 melanomas. Am J Surg Pathol 2015; 39: 1197-205. PubMed
- 83. Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small surprise: small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol 2014; 233: 209-14. ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Mixed neuroendocrine-non-neuroendocrine neoplasm (MiNEN) of the cervix in a 38-year-old female: a case report and review of literature
Josh Matthew B. Chen, Denise B. Andal, Benedict Jose P. Canora, Claire Anne Therese M. Hemedez
Human Pathology Reports.2026; 43: 300815. CrossRef - Neuroendocrine Neoplasms of the Gastrointestinal Tract: Morphology, WHO 2022 Grading, and Prognostic Perspectives
Hussein Qasim, Shaima' Dibian, Mohammad Abu Shugaer, Karis Khattab, Mudhaffer Touqan, Matteo Luigi Giuseppe Leoni , Giustino Varrassi
Cureus.2026;[Epub] CrossRef - When Immunophenotype Is Not Identity: A Clinicopathological Review of Neuroendocrine Differentiation in Tumors of the Female Genital Tract
Catalin-Bogdan Satala, Alina-Mihaela Gurau, Gabriela Patrichi, Roxana-Cristina Mehedinti, Andy Radu Leibovici, Gabriela Gurau
Diagnostics.2026; 16(10): 1573. CrossRef - A rare case report of primary ovarian carcinoid presenting with constipation
Xiaofeng Deng, Qian Huang, Bangfang Xie, Hailong Huang, Jianguo Chen
Frontiers in Oncology.2025;[Epub] CrossRef - Clinical, pathological characteristics, and therapeutic outcomes of primary ovarian carcinoid tumors: a case series of 15 cases
Xinyue Dai, Suidan Chen, Simeng Yang
World Journal of Surgical Oncology.2025;[Epub] CrossRef - Smart Red Blood Cell Carriers: A Nanotechnological Approach to Cancer Drug Delivery
Ioannis Tsamesidis, Georgios Dryllis, Sotirios P. Fortis, Andreas Sphicas, Vasiliki Konstantinidou, Maria Chatzidimitriou, Stella Mitka, Maria Trapali, Petros Skepastianos, Anastasios G. Kriebardis, Ilias Pessach
Current Issues in Molecular Biology.2025; 47(9): 711. CrossRef - Imaging of Gynecologic Neuroendocrine Tumors: A Case-Based Pictorial Essay
Ana Paula Bavaresco, Ulysses S. Torres, Mayara S. Cruz, Vitor V.C. Machado, Cynthia L.P. Borborema, Giovanna S. Torre, Jhonata Soares Da Silva, Tulio A. Kawai, Gustavo R.A. Focchi, Eduardo O. Pacheco, Aley Talans, Daniel Bekhor, Ana Paula C. Moura, Lucas
Seminars in Ultrasound, CT and MRI.2025;[Epub] CrossRef - Challenges in Diagnosis and Management of Ovarian Neuroendocrine Carcinoma: A Case of Aggressive Disease With Multimodal Treatment Approach
Javeria Haider, Humera Mahmood, Muhammad Faheem, Shaista Khurshid, Abdullah, Biruk Demisse Ayalew, Humza Saeed
Clinical Case Reports.2025;[Epub] CrossRef - Neuroendocrine Marker Expression in Primary Non-neuroendocrine Epithelial Tumors of the Ovary: A Study of 551 Cases
Michaela Kendall Bártů, Kristýna Němejcová, Romana Michálková, Quang Hiep Bui, Jana Drozenová, Pavel Fabian, Oluwole Fadare, Jitka Hausnerová, Jan Laco, Radoslav Matěj, Gábor Méhes, Adam Šafanda, Naveena Singh, Petr Škapa, Zuzana Špůrková, Simona Stolnicu
International Journal of Gynecological Pathology.2024; 43(2): 123. CrossRef - Diagnostic and therapeutic challenge of neuroendocrine endometrial carcinoma: a case report
Hariyono Winarto, David Calvin, Fitriyadi Kusuma, Kartiwa Hadi Nuryanto, Yuri Feharsal, Dewita Nilasari, Hartono Tjahjadi
The Pan African Medical Journal.2024;[Epub] CrossRef - Neuroendocrine carcinoma of ovary: Hitherto rare entity in primary ovarian tumors
Md A. Osama, Seema Rao, Punita Bhardwaj, Geeta Mediratta, Sunita Bhalla, Sonia Badwal
Indian Journal of Pathology and Microbiology.2023; 66(4): 855. CrossRef - Mixed neuroendocrine–non-neuroendocrine neoplasm with mucinous adenocarcinoma and amphicrine carcinoma components in the bile duct: an autopsy case
Toji Murabayashi, Yoshihide Kanno, Takashi Odaira, Shinsuke Koshita, Takahisa Ogawa, Hiroaki Kusunose, Toshitaka Sakai, Keisuke Yonamine, Kazuaki Miyamoto, Fumisato Kozakai, Kazuki Endo, Yutaka Noda, Takashi Sawai, Kei Ito
Clinical Journal of Gastroenterology.2023; 16(2): 310. CrossRef - Coexistence of Papillary Thyroid Carcinoma and Strumal Carcinoid Arising from Struma Ovarii in Pregnant Women: a Case Report and Review
Myungsoo Im, Doohwa Kim, Soree Ryang, Bo Hyun Kim
International Journal of Thyroidology.2023; 16(1): 134. CrossRef - Role of radiotherapy in the management of rare gynaecological cancers
R. Morcet-Delattre, S. Espenel, P. Tas, C. Chargari, A. Escande
Cancer/Radiothérapie.2023; 27(8): 778. CrossRef - Small cell carcinoma of the ovary, pulmonary type: A role for adjuvant radiotherapy after carboplatin and etoposide?
Anase S. Asom, Ricardo R. Lastra, Yasmin Hasan, Lori Weinberg, Gini F. Fleming, Katherine C. Kurnit
Gynecologic Oncology Reports.2022; 39: 100925. CrossRef - MicroRNA and Metabolic Profiling of a Primary Ovarian Neuroendocrine Carcinoma Pulmonary-Type Reveals a High Degree of Similarity with Small Cell Lung Cancer
Stefano Miglietta, Giulia Girolimetti, Lorena Marchio, Manuela Sollazzo, Noemi Laprovitera, Sara Coluccelli, Dario De Biase, Antonio De Leo, Donatella Santini, Ivana Kurelac, Luisa Iommarini, Anna Ghelli, Davide Campana, Manuela Ferracin, Anna Myriam Perr
Non-Coding RNA.2022; 8(5): 64. CrossRef - Neuroendocrine Carcinomas of the Uterine Cervix, Endometrium, and Ovary Show Higher Tendencies for Bone, Brain, and Liver Organotrophic Metastases
Hyung Kyu Park
Current Oncology.2022; 29(10): 7461. CrossRef - Uterine carcinoma admixed with neuroendocrine carcinoma
Maria Victoria Olinca, Anca Potecă, Elvira Brătilă, Mihai Mitran
Ginecologia.ro.2022; 4(38): 32. CrossRef - The puzzle of gynecologic neuroendocrine carcinomas: State of the art and future directions
Giuseppe Caruso, Carolina Maria Sassu, Federica Tomao, Violante Di Donato, Giorgia Perniola, Margherita Fischetti, Pierluigi Benedetti Panici, Innocenza Palaia
Critical Reviews in Oncology/Hematology.2021; 162: 103344. CrossRef - Pitfalls and challenges in managing neuroendocrine carcinoma of gynecological origin: A case series and brief review
Lauren E. Farmer, Rutmi U. Goradia, Nisha A. Lakhi
Clinical Case Reports.2021;[Epub] CrossRef - Primary mixed large cell neuroendocrine and high grade serous carcinoma of the endometrium
Liesel Elisabeth Hardy, Zia Chaudry, King Wan, Chloe Ayres
BMJ Case Reports.2020; 13(9): e234977. CrossRef - Neuroendocrine carcinoma of the endometrium: Disease course, treatment, and outcomes
Kathryn Schlechtweg, Ling Chen, Caryn M. St. Clair, Ana I. Tergas, Fady Khoury-Collado, June Y. Hou, Alexander Melamed, Alfred I. Neugut, Dawn L. Hershman, Jason D. Wright
Gynecologic Oncology.2019; 155(2): 254. CrossRef - Peritoneal Fluid Cytology of Disseminated Large Cell Neuroendocrine Carcinoma Combined with Endometrioid Adenocarcinoma of the Endometrium
Yong-Moon Lee, Min-Kyung Yeo, Song-Yi Choi, Kyung-Hee Kim, Kwang-Sun Suh
Journal of Pathology and Translational Medicine.2019; 53(6): 407. CrossRef - Pro-Gastrin Releasing Peptide: A New Serum Marker for Endometrioid Adenocarcinoma
Mine Kiseli, Gamze Sinem Caglar, Asli Yarci Gursoy, Tolga Tasci, Tuba Candar, Egemen Akincioglu, Emre Goksan Pabuccu, Nurettin Boran, Gokhan Tulunay, Haldun Umudum
Gynecologic and Obstetric Investigation.2018; 83(6): 540. CrossRef - Tumeur neuroendocrine à petite cellule de l’endomètre : prise en charge originale
E. Galmiche, N. Hudry, P. Sagot, P. Ginod, S. Douvier
Gynécologie Obstétrique Fertilité & Sénologie .2017; 45(6): 381. CrossRef - Twist on a classic: vitamin D and hypercalcaemia of malignancy
Juan C Osorio, Masha G Jones, Nina Schatz-Siemers, Stephanie J Tang
BMJ Case Reports.2017; 2017: bcr-2017-220819. CrossRef - Mixed Neuroendocrine-Nonneuroendocrine Neoplasms (MiNENs): Unifying the Concept of a Heterogeneous Group of Neoplasms
Stefano La Rosa, Fausto Sessa, Silvia Uccella
Endocrine Pathology.2016; 27(4): 284. CrossRef - Neuroendocrine tumours in rare sites: differences in nomenclature and diagnostics—a rare and ubiquitous histotype
Elia Guadagno, Gaetano De Rosa, Marialaura Del Basso De Caro
Journal of Clinical Pathology.2016; 69(7): 563. CrossRef - Primary ovarian neuroendocrine tumor arising in association with a mature cystic teratoma: A case report
Nicolas M. Orsi, Mini Menon
Gynecologic Oncology Reports.2016; 17: 83. CrossRef - Benign Endometrial Polyp and Primary Endometrial Small Cell Neuroendocrine Carcinoma Confined to the Polyp: A Rare Association
Pembe Oltulu, Ceyhan Uğurluoğlu, Ayşenur Uğur, Sıdıka Fındık, Lema Tavlı
Journal of Clinical and Experimental Investigations.2016;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
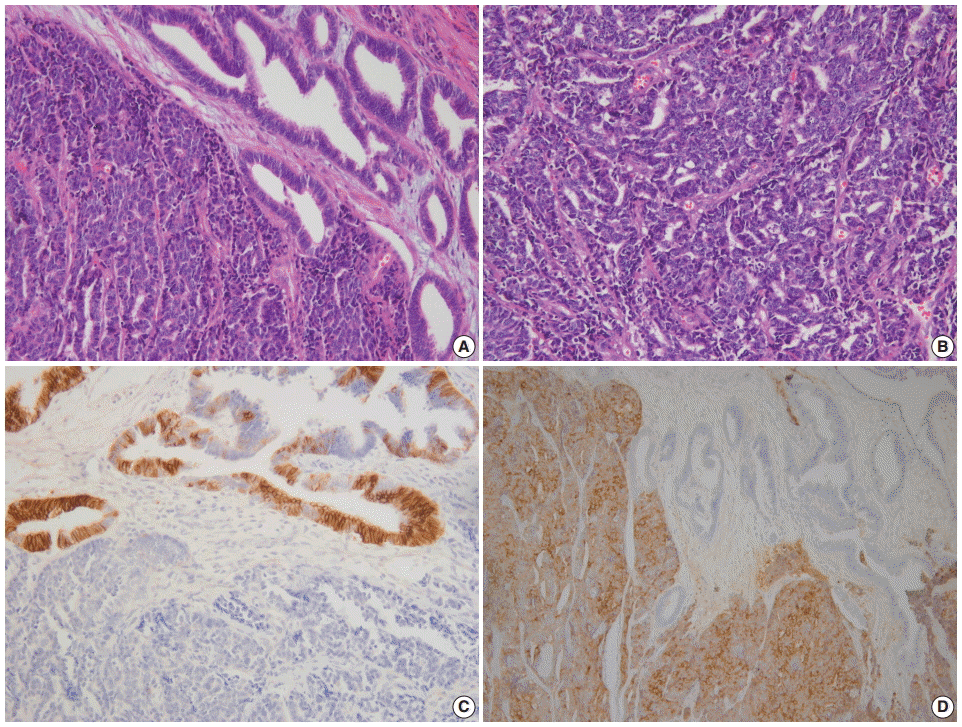
Neuroendocrine Tumors of the Female Reproductive Tract: A Literature Review



Fig. 1. (A) Endometrial large cell neuroendocrine carcinoma (LCNEC) admixed with grade 1 endometrioid carcinoma. (B) Large vesicular nuclei and prominent nucleoli in LCNEC. (C) CD56 immunostaining: positive in endometrioid carcinoma and negative in LCNEC. (D) Diffuse synaptophysin expression in LCNEC.
Fig. 2. (A) Dedifferentiated carcinoma composed of undifferentiated carcinoma and grade 1 endometrioid carcinoma. (B) Dyscohesive tumor cells growing in a patternless fashion without gland formation. Focal positivity of cytokeratin (C) and synaptophysin (D) immunostaining.
Fig. 3. (A) Grade 3 endometrioid carcinoma mimicking neuroendocrine carcinoma at low magnification. (B) At high magnification, grade 3 endometrioid carcinoma shows similar tumor cells in the solid and glandular areas. Focal positivity for neuroendocrine markers of CD56 (C) and synaptophysin (D).
Fig. 1.
Fig. 2.
Fig. 3.
Neuroendocrine Tumors of the Female Reproductive Tract: A Literature Review

