 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 50(6); 2016 > Article
-
Brief Case Report
Goblet Cell Carcinoid of the Rectum in a Patient with Neurofibromatosis Type 1 - Youngjin Kang, Jung-Woo Choi,, Younghye Kim, Hwa Eun Oh, Ju-Han Lee, Young-Sik Kim
-
Journal of Pathology and Translational Medicine 2016;50(6):482-485.
DOI: https://doi.org/10.4132/jptm.2016.02.27
Published online: May 29, 2016
Department of Pathology, Korea University Ansan Hospital, Ansan, Korea
- Corresponding Author Jung-Woo Choi, MD, PhD Department of Pathology, Korea University Ansan Hospital, 123 Jeokgeum-ro, Danwon-gu, Ansan 15355, Korea Tel: +82-31-412-6753, Fax: +82-31-412-5324, E-mail: nausika@korea.ac.kr
• Received: January 27, 2016 • Revised: February 24, 2016 • Accepted: February 27, 2016
© 2016 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
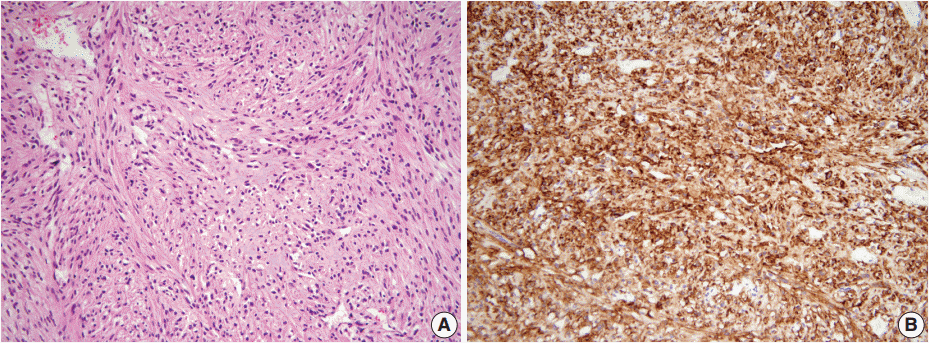
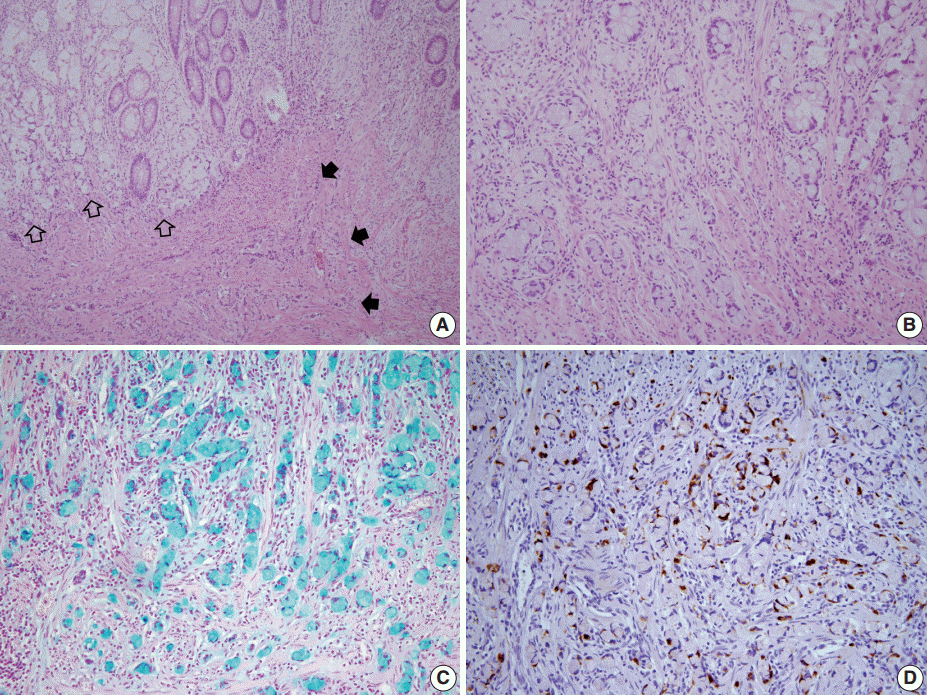
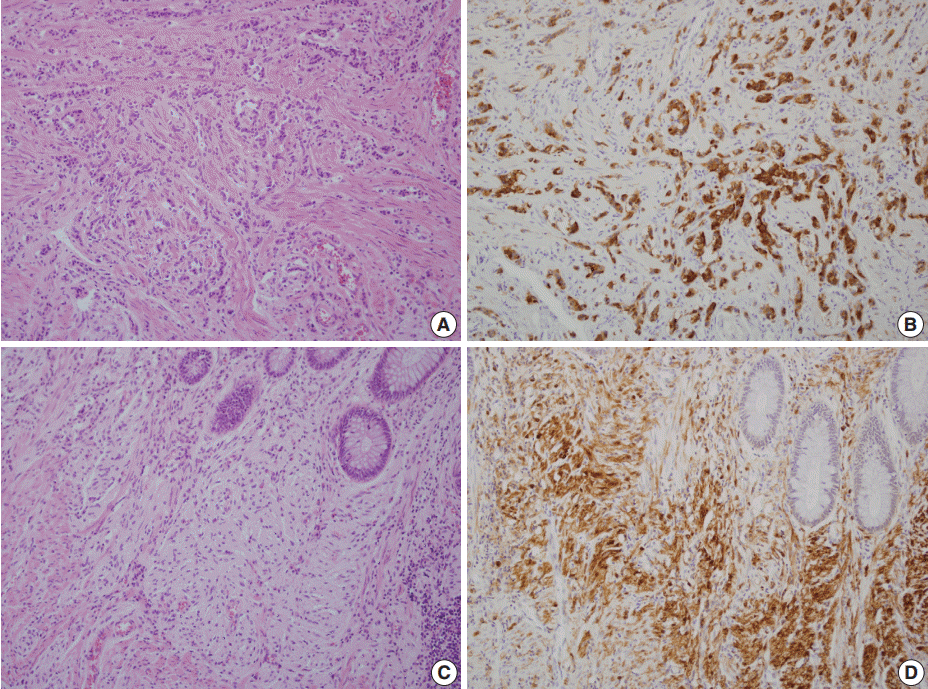
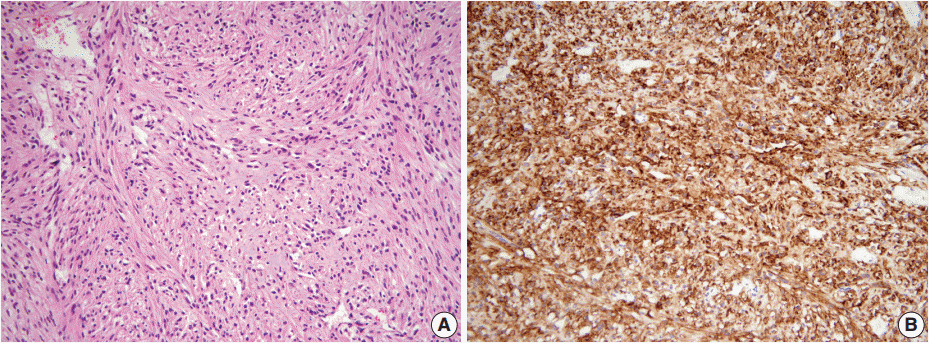
- A 46-year-old woman who congenitally had clinical features of NF-1 such as multiple neurofibromas of skin and café au lait macule has suffered from recurrent NETs in rectum since she received an operation for ovarian mucinous cystadenocarcinoma in 2010. The ovarian tumor showed neither recurrence nor distant metastasis during follow-up. She denied all the symptoms indicative of gastrointestinal diseases or carcinoid syndrome. Upon colonoscopy, the rectal mucosa revealed multifocal whitish patches, multiple biopsies from which revealed a coincidental occurrence of GCC, NETs, and neurofibroma. The abdominal computerized tomographic scan incidentally detected a submucosal mass in the jejunum. She underwent a low anterior resection of rectum and a wedge resection of jejunum. During the operation, the appendix was unremarkable by inspection. Grossly, the involved segment of rectum was about 10 cm in length and contained four to five patchy indurated lesions measuring 5.2×3.5 cm across in the largest one, which were limited to the mucosa and submucosa. Microscopically, the tumor consisted of small nests of signet-ring-like goblet cells (Fig. 1A, B), which positively stained with alcian blue pH 2.5 (Fig. 1C). Immunohistochemical stains for CD56, synaptophysin (Fig. 1D), and chromogranin A revealed scattered neuroendocrine cells, leading to the diagnosis of rectal GCC. Adjacent to rectal GCC, there were multifocal minute grade 1 NETs according to the grading system of 2010 World Health Organization (WHO) classification of NETs (Fig. 2A, B) [2]. Neurofibromatosis in the background of GCC and NETs was confirmed by S-100 protein immunostain (Fig. 2C, D). The submucosal tumor in the jejunum was composed of bland-looking spindle cells which expressed both c-kit and CD34 by immunohistochemistry, and diagnosed as very low risk GIST (size, 1.0 cm; mitotic count, <1/50 high power fields) (Fig. 3A, B). Ten months after the operation, the patient was alive without recurrence or metastasis.
CASE REPORT
- Even though GCC has been regarded as a mucin-producing NETs exclusively arising in the appendix [3], it is unsettled whether GCC should be classified as part of NETs or as variants of adenocarcinoma. The different histogenesis, the relative paucity of neuroendocrine cells, and more aggressive behavior have made GCC look different from NETs. It has been supposed that the dual neuroendocrine and intestinal differentiation in GCC emerge from undifferentiated stem cells at the base of the intestinal crypts, whereas NETs initiate from subepithelial neuroendocrine cells in the mucosa [4]. Moreover, the neuroendocrine cells of GCC are considerably fewer than those of NETs. As GCC has been more aggressive than NETs, adjuvant therapy after resection could be attempted commonly. Nevertheless, the genetic resemblance of GCC to NETs has precluded its being categorized as a subtype of adenocarcinoma. Indeed, GCC had allelic losses of 11q, 16q, or 18q which are frequent in gastrointestinal NETs, whereas KRAS, DPC4, or CTNNB1 mutations signaling for colorectal adenocarcinoma were undiscovered [5]. The co-occurrence of GCC and NETs in this case might also implicate the close oncogenic relationship between them.
- The rectum is an unusual site for GCC, and less than 10 cases of GCC arising in the rectum have been reported [6]. Among them, there have been no patients with NF-1 as the present case, while a NF-1 patient with appendiceal GCC was recently described [7]. Somatic inactivation of the NF1 has been the molecular hallmark of NF-1. A recent analysis has demonstrated that unlike sporadic GISTs, NF-1–related GISTs did not have activating mutations of KIT or PDGFRA, but showed somatic inactivation of NF1 more frequently [8]. In addition, the tumor cells of gastric NET in a NF-1 patient showed germline nonsense mutation and loss of heterozygosity of NF1 [9]. Therefore, the occurrence of GCC along with NET, GIST and neurofibromatosis in this patient suggest that GCC might be one of NF-1–related tumors, and might have arisen from NF1 gene dysregulation. However, the genetic association between GCC and NF-1 needs to be established further.
- Rectal GCC should be differentiated from metastasis of appendiceal GCC and from signet ring cell carcinoma, which usually forms luminal space and is negative for neuroendocrine markers by immunohistochemistry. Although the present patient had ovarian mucinous cystadenocarcinoma, the microscopic features between the tumors of rectum and ovary were obviously different.
- In conclusion, for the first time we report rectal GCC arising in a NF-1 patient. This case will contribute to broadening the scope of gastrointestinal tumors, which NF-1 patients can harbor. More data need to be accumulated to determine the biologic characteristics and the optimal treatment strategies for this rare tumor.
DISCUSSION
Acknowledgments
Fig. 1.The rectum shows a combined occurrence of goblet cell carcinoid (GCC, empty arrows) and neuroendocrine tumors (filled arrows) as well as neurofibromatosis (A). GCC is mostly composed of small nests of signet-ring-like goblet cells (B), and the cytoplasmic mucin is readily identified with alcian blue pH 2.5 (C). (D) Synaptophysin immunostain clearly shows a few scattered neuroendocrine cells in GCC.
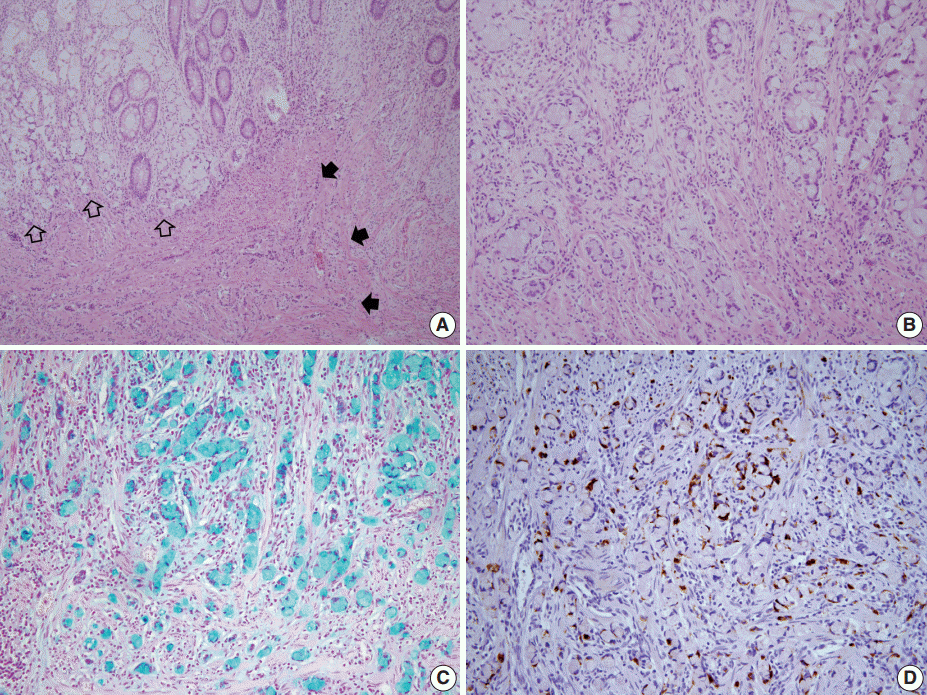
Fig. 2.Neuroendocrine tumors (NETs) consist of small uniform cells which exclusively form trabecular structures (A) and diffusely stained with synaptophysin (B). In the vicinity of both goblet cell carcinoid and NETs, proliferation of slender spindle cells morphologically compatible with neurofibromatosis (C) is evident with S-100 protein immunostain (D).
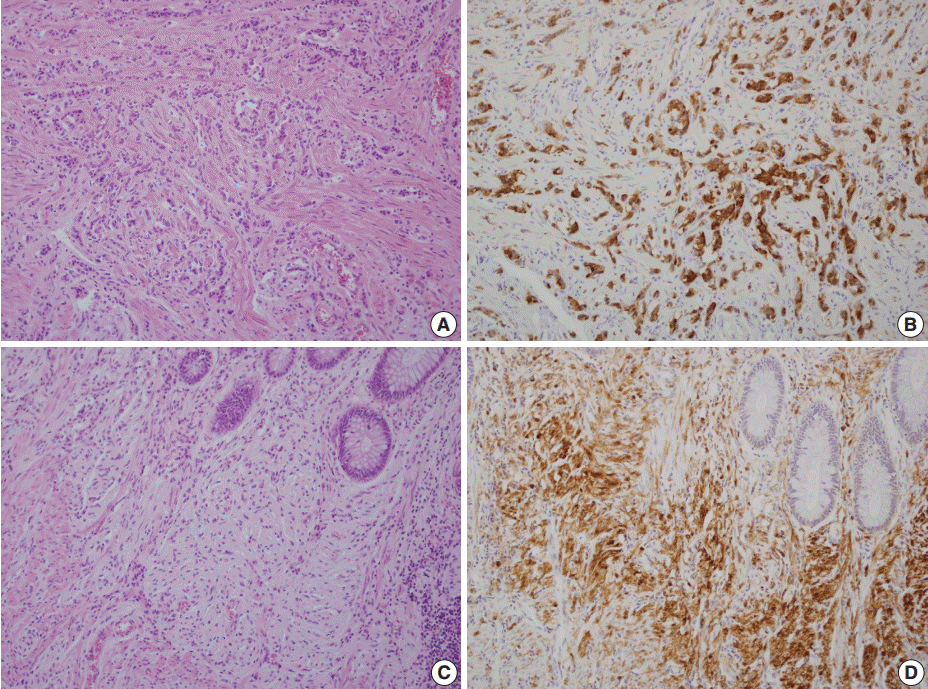
Fig. 3.The submucosal tumor in the jejunum is composed of bland-looking spindle cells (A) which highly expressed c-Kit by immunohistochemistry (B).
- 1. Agaimy A, Vassos N, Croner RS. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol 2012; 5: 852-62. PubMedPMC
- 2. Rindi G, Klimstra DS, Arnold R, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system. 4th ed. Lyon: IARC Press, 2010; 13-4.
- 3. Gui X, Qin L, Gao ZH, Falck V, Harpaz N. Goblet cell carcinoids at extraappendiceal locations of gastrointestinal tract: an underrecognized diagnostic pitfall. J Surg Oncol 2011; 103: 790-5. ArticlePubMedPDF
- 4. Hofler H, Kloppel G, Heitz PU. Combined production of mucus, amines and peptides by goblet-cell carcinoids of the appendix and ileum. Pathol Res Pract 1984; 178: 555-61. ArticlePubMed
- 5. Stancu M, Wu TT, Wallace C, Houlihan PS, Hamilton SR, Rashid A. Genetic alterations in goblet cell carcinoids of the vermiform appendix and comparison with gastrointestinal carcinoid tumors. Mod Pathol 2003; 16: 1189-98. ArticlePubMed
- 6. Yamabuki T, Omi M, Yonemori A, et al. Goblet cell carcinoid of the rectum with lymph node metastasis: report of a case. Surg Today 2011; 41: 1284-9. ArticlePubMedPDF
- 7. Gregersen T, Holt N, Gronbaek H, Vogel I, Jørgensen LJ, Krogh K. Goblet cell carcinoid in a patient with neurofibromatosis type 1: a rare combination. Case Rep Gastrointest Med 2012; 2012: 185730.ArticlePubMedPMCPDF
- 8. Maertens O, Prenen H, Debiec-Rychter M, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006; 15: 1015-23. PubMed
- 9. Stewart W, Traynor JP, Cooke A, et al. Gastric carcinoid: germline and somatic mutation of the neurofibromatosis type 1 gene. Fam Cancer 2007; 6: 147-52. PubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- A rare goblet cell adenocarcinoma arising from Barrett’s esophagus: the first reported case in the esophagus
Chi Eun Oh, Sung Eun Kim, Sun-Ju Oh
Journal of Pathology and Translational Medicine.2024; 58(2): 81. CrossRef - Goblet cell adenocarcinoma of the anal canal with perianal Paget disease: A rare case report with literature review
Minhua Li, Xiaofei Yao
Medicine.2023; 102(16): e33598. CrossRef - A Case of Goblet Cell Carcinoid of the Transverse Colon
Yuko Kamata, Hiroshi Kuwabara, Masafumi Akasu, Morio Koike
Nippon Daicho Komonbyo Gakkai Zasshi.2022; 75(7): 366. CrossRef - Mixed Neuroendocrine Non-neuroendocrine Neoplasm of Anorectum with Goblet Cell Morphology
Sandhya Biswal, Anirban Kundu, Ankit Sahoo, Prakash Kumar Sasmal, Biswajit Sahoo, Suvradeep Mitra
Journal of Gastrointestinal Cancer.2021; 52(3): 1093. CrossRef - Goblet cell carcinoid of the rectum: a case report
Yoshiyuki Inoue, Hisanaga Horie, Yuko Homma, Ai Sadatomo, Makiko Tahara, Koji Koinuma, Hironori Yamaguchi, Toshiki Mimura, Atsushi Kihara, Alan Kawarai Lefor, Naohiro Sata
Surgical Case Reports.2020;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
Goblet Cell Carcinoid of the Rectum in a Patient with Neurofibromatosis Type 1



Fig. 1. The rectum shows a combined occurrence of goblet cell carcinoid (GCC, empty arrows) and neuroendocrine tumors (filled arrows) as well as neurofibromatosis (A). GCC is mostly composed of small nests of signet-ring-like goblet cells (B), and the cytoplasmic mucin is readily identified with alcian blue pH 2.5 (C). (D) Synaptophysin immunostain clearly shows a few scattered neuroendocrine cells in GCC.
Fig. 2. Neuroendocrine tumors (NETs) consist of small uniform cells which exclusively form trabecular structures (A) and diffusely stained with synaptophysin (B). In the vicinity of both goblet cell carcinoid and NETs, proliferation of slender spindle cells morphologically compatible with neurofibromatosis (C) is evident with S-100 protein immunostain (D).
Fig. 3. The submucosal tumor in the jejunum is composed of bland-looking spindle cells (A) which highly expressed c-Kit by immunohistochemistry (B).
Fig. 1.
Fig. 2.
Fig. 3.
Goblet Cell Carcinoid of the Rectum in a Patient with Neurofibromatosis Type 1

