 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 48(6); 2014 > Article
-
Brief Case Report
Peritoneal Carcinosarcoma and Ovarian Papillary Serous Carcinoma Are the Same Origin: Analysis of TP53 Mutation and Microsatellite Suggests a Monoclonal Origin - Chang Gok Woo, Dae Shik Suh1, Joo Young Kim, Chang Ohk Sung, Jene Choi, Kyu-Rae Kim,
-
Korean Journal of Pathology 2014;48(6):449-453.
DOI: https://doi.org/10.4132/KoreanJPathol.2014.48.6.449
Published online: December 31, 2014
Departments of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
1Departments of Obstetrics and Gynecology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding Author: Kyu-Rae Kim, M.D. Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 138-736, Korea Tel: +82-2-3010-4514, Fax: +82-2-472-7898, E-mail: 'krkim@amc.seoul.kr'
• Received: December 27, 2013 • Revised: February 20, 2014 • Accepted: February 21, 2014
© 2014 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- The patient, a 57-year-old postmenopausal woman, was found to have a palpable mass in the right lower quadrant without other clinical symptoms. Her past medical history was unremarkable. On computerized tomographic scan, multiple heterogeneously enhanced masses of variable size with lobulating contours were identified in the abdominal and pelvic cavities. Whole body 18F-fluorodeoxyglucose positron emission tomography scan revealed multiple hypermetabolic masses in the bilateral paracolic gutter, mesentery, and pelvic cavity. Of the tumor markers, the serum level of cancer antigen (CA) 72-4 was increased (5.7 U/mL; normal range, 0 to 4 U/mL), but CA 19-9 (11.0 U/mL) and carcinoembyonic antigen (1.5 ng/mL) were within normal range.
- Presuming a sarcoma of an unknown primary site, a needle biopsy was performed on the paracolic mass for pathological diagnosis and determination of the primary site. The mass was determined to be carcinosarcoma (a malignant mixed Müllerian tumor). Total abdominal hysterectomy, bilateral salpingo-oo-phorectomy, right hemicolectomy, and low anterior resection were performed. Currently, the patient received six cycles of postoperative chemotherapy composed of carboplatin and paclitaxel, and she has survived without tumor recurrence for eight months postoperatively. However, a vesicovaginal fistula developed during the treatment as a complication of the surgery.
- Grossly, the largest mass (12 cm in diameter) was located in the pericolic soft tissue of the ascending colon. The mass protruded into the peritoneal cavity and displaced the colon to the anterior side, but it was well delineated from the colonic wall without invasion. The cut surface of the pericolic mass was bluish pink and firm, and it also had focal necrotic areas. The ovaries were slightly enlarged bilaterally, with the right ovary measuring 5×4×3.8 cm and the left ovary measuring 2×1.5×1.5 cm. Both ovaries had been entirely replaced by solid, well-defined and lobulated tumor tissue. The cut surfaces of both ovaries were yellowish white, firm, and granular. A separate, well-defined, multilobulated mass was also identified within the perirectal soft tissue. This mass measured 3.8 cm in diameter and did not invade the rectal wall.
- Microscopically, the tumor in the pericolic soft tissue showed characteristic features of carcinosarcoma and contained both carcinomatous and sarcomatous components. The carcinomatous portion showed histopathologic features of a high-grade papillary serous carcinoma, with complex and interconnecting papillary structures lined by stratified malignant epithelial cells. The sarcomatous section was composed of malignant spindle cells showing nuclear atypia, increased cellularity, a high mitotic index, and occasional malignant-appearing cartilage (Fig. 1A). The bilateral ovarian and perirectal tumors were histologically characterized by complex and interconnecting papillary structures lined by stratified malignant epithelial cells showing marked cytologic atypia, a high nuclear-to-cytoplasmic ratio and frequent mitotic structures (Fig. 1B). A sarcomatous component was not found despite a thorough examination of the bilateral ovarian tumors and the perirectal mass. A metastatic mass identified in a pelvic lymph node was entirely carcinomatous.
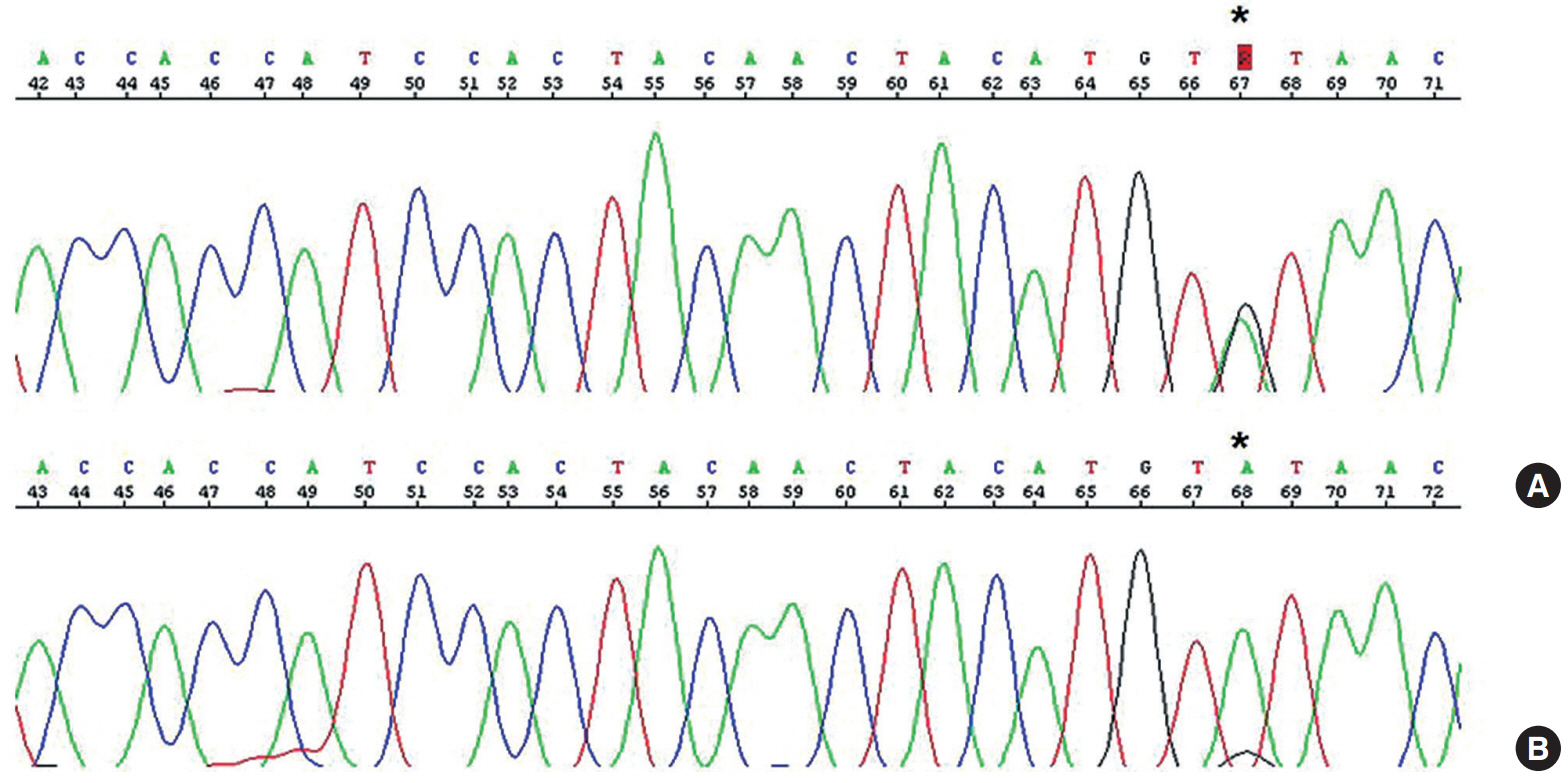
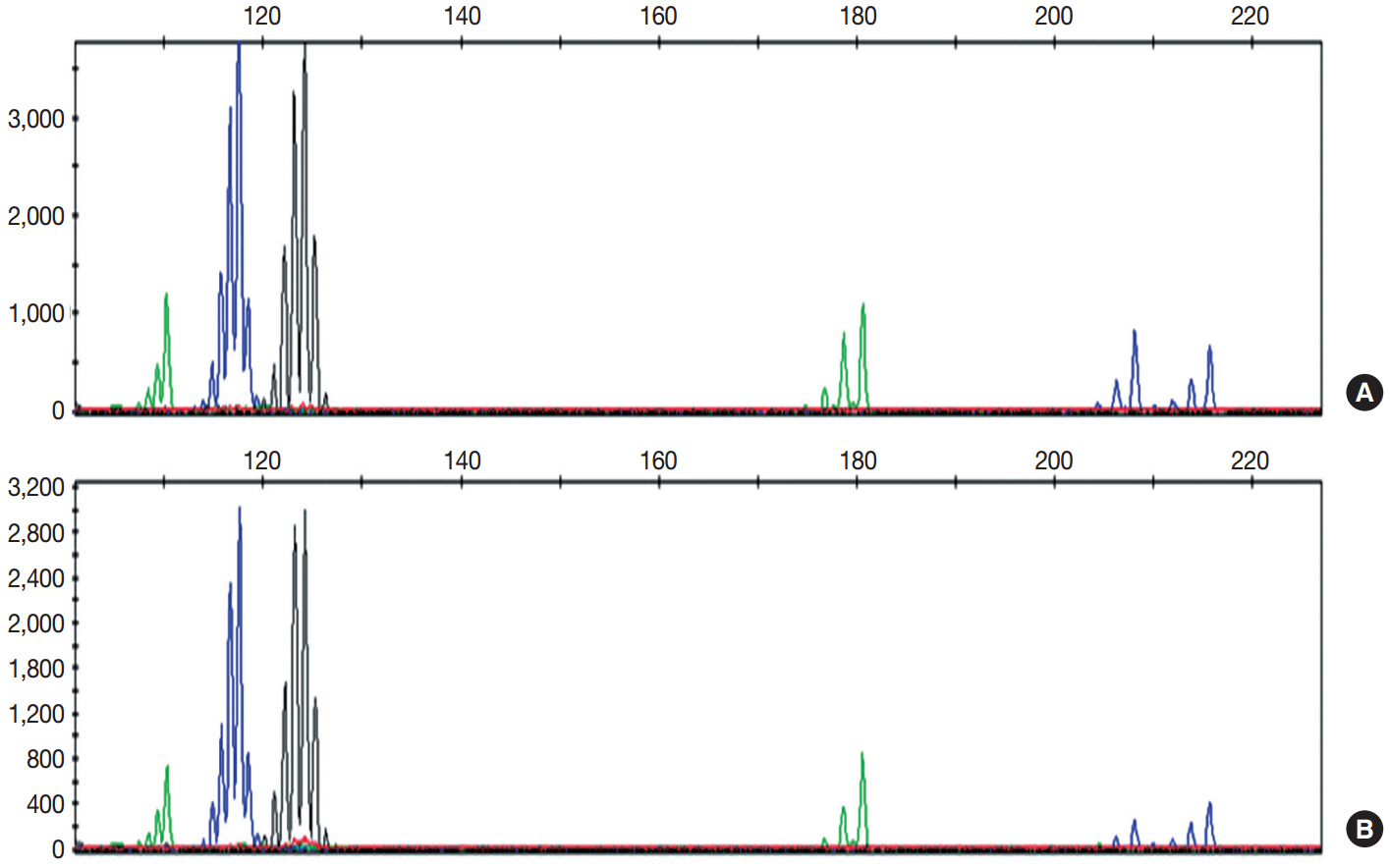
- On immunohistochemical staining, both the carcinomatous and the sarcomatous components of the pericolic mass and the carcinomatous component of the ovarian and perirectal masses showed strong immunopositivity for p53 (1:1,500, clone DO-7, Dako, Glostrup, Denmark) (Fig. 1C, D). For the molecular genetic analysis, formalin-fixed, paraffin-embedded tissue was obtained from the pericolic and right ovarian masses, which showed characteristic features of carcinosarcoma and high-grade papillary serous carcinoma, respectively. An area of the tumor in which the percentage of tumor cells exceeded a minimum of 50% of all cells was marked and scraped with a knife blade. Genomic DNA was prepared by proteinase K digestion and followed by phenol/chloroform extraction. Polymerase chain reaction covering the coding region of exons 5, 6, 7, and 8 of TP53 was performed, and the amplified DNA was subjected to direct sequencing by the Sanger method (Applied Biosystems, Foster City, CA, USA) and compared with genomic repository data to determine the mutational status of TP53. An identical TP53 somatic point mutation (c.G713A; p.C238Y) in exon 7 was detected in both tumors (Fig. 2). Additionally, microsatellite instability (MSI) was assessed at the loci BAT25, BAT26, D2S123, D5S346, and D17S250. We selected a consensus panel of five microsatellite markers, BAT25, BAT26, D5S346, D17S250, and D2S123, which have been used to diagnose the MSI status of hereditary non-polyposis colorectal cancer (HNPCC) based on the Bethesda guidelines. All of these markers are highly polymorphic microsatellite markers (Fig. 3). All tested microsatellite markers had identical DNA profile patterns with the same allelic sizes between the tumors of the two areas. The overall probability that all five microsatellite markers were identical between the two different tumors was extremely low.
CASE REPORT
- A malignant tumor showing a mixed histology with both carcinomatous and sarcomatous components can arise in almost every organ in the body. In the pelvic organs, carcinosarcoma usually occurs in the uterus, but it can also arise in the ovary, fallopian tube, broad ligament, mesentery, cul-de-sac, and pelvic peritoneum, and it comprises approximately 1%–4% of all ovarian malignancies. Currently, it is thought to be a form of metaplastic carcinoma [1] because 1) pure carcinomatous foci are often identified on extensive sampling of the carcinosarcoma, 2) identical genetic changes are identified in both the carcinomatous and the sarcomatous components, 3) carcinosarcoma is often preceded by or associated with endometrioid or serous carcinoma, and 4) the response to adjuvant therapy against highgrade carcinoma is more effective than sarcoma-based therapy [2].
- The pathogenetic mechanism of peritoneal carcinosarcoma has not been completely clarified. Since carcinosarcoma is often associated with synchronous or metachronous gynecologic tumors of Mullerian origin, including serous and endometrioid carcinoma [3,4], and some extrauterine carcinosarcomas were found to be associated with endometriosis or Wolffian duct remnants, carcinosarcoma is thought to arise from extraovarian endometriosis [5], immature multipotential cells of the surface epithelium or subcapsular connective tissue of the ovary. The peritoneal lining epithelium in the broad ligament, mesentery, cul-de-sac, and pelvic peritoneum also have the potential to give rise to Mullerian tumors because the Mullerian ducts are derived from the longitudinal invaginations of the mesothelium, which gives rise to the peritoneal lining. Therefore, theoretically, extragenital carcinosarcoma may occur as a primary neoplasm from the pelvic peritoneum.
- Since there has been no effective treatment modality for carcinosarcoma, efforts have been made to identify underlying molecular mechanisms to make use of targeted therapies [6]. Carcinosarcomas in different organs are associated with different oncogenic mutations, including mutations in the epidermal growth factor receptor (EGFR) gene, the PIK3CA gene, and the p53 gene in tumors of the lung, uterus, and ovary, respectively, indicating distinct oncogenic mechanisms for carcinosarcomas in these different organs. Loss of nuclear WT1 expression in the mesenchymal component might be involved in sarcomatous differentiation. Our molecular analysis revealed an identical mutation in TP53 occurring in both the carcinomatous and sarcomatous components. All five microsatellites of two tumors retained very similar migration patterns, and a main peak of each microsatellite marker was seen at the same length. These findings of molecular studies suggest that these components share a monoclonal origin [7]. Moreover, one case of extragenital carcinosarcoma in the literature had the same mutation as a preexisting ovarian epithelial carcinoma [8]. The results of our molecular studies in this case and in previous reports support that the ovarian carcinoma and peritoneal carcinosarcoma had the same origin.
- Although the carcinosarcoma and the pure carcinoma share a common origin, however, the clinical significance of carcinosarcoma is significantly different from that of pure carcinoma. Patients with ovarian carcinosarcoma had worse five year progression-free and disease-specific survival rates than ovarian papillary serous carcinoma patients at the same International Federation of Gynecologists and Oncologists (FIGO) stages. Patients with a heterologous sarcomatous component also had a worse prognosis than those with a homologous component, although the difference was not statistically significant. The objective response rate to platinum-based chemotherapy was found to be significantly lower in patients with carcinosarcoma compared to those with serous carcinoma.
- In conclusion, in cases having two separate tumors in the same patient at distant locations with different histologic findings, meticulous histological examination is necessary, and molecular studies can be helpful to identify the origin of the tumors.
DISCUSSION
Fig. 1.The pericolic mass shows both carcinomatous and sarcomatous components composed of complex and interconnecting papillary structures lined by stratified malignant epithelial cells and a malignant cartilaginous element (A), but the ovarian mass contains only a carcinomatous component (B). On immunohistochemical staining, both the carcinomatous and the sarcomatous components in the pericolic mass (C) and the carcinomatous component of the ovarian tumor (D) show strong immunopositivity for p53.

Fig. 2.An identical somatic point mutation (TGT to TAT; p.C238Y) is detected in exon 7 of TP53 in both the carcinomatous and the sarcomatous component of the pericolic tumor mass (A) and in the epithelial component of the ovarian tumor (B).

Fig. 3.Several peaks show the same migration pattern, and each main peak of the five markers is seen at the same migration length in the pericolic tumor mass (A) and the epithelial component of the ovarian tumor (B).

- 1. McCluggage WG. Uterine carcinosarcomas (malignant mixed Mullerian tumors) are metaplastic carcinomas. Int J Gynecol Cancer 2002; 12: 687–90. ArticlePubMed
- 2. van Rijswijk RE, Tognon G, Burger CW, Baak JP, Kenemans P, Vermorken JB. The effect of chemotherapy on the different components of advanced carcinosarcomas (malignant mixed mesodermal tumors) of the female genital tract. Int J Gynecol Cancer 1994; 4: 52–60. ArticlePubMed
- 3. Ma CJ, Yang SF, Huang CC, et al. Malignant mixed mullerian tumor of primary mesenteric origin associated with a synchronous ovarian cancer: case report and literature review. Eur J Gynaecol Oncol 2008; 29: 289–93. PubMed
- 4. Handa Y, Kato H, Kaneuchi M, Saitoh Y, Yamashita K. High-grade broad ligament cancer of mullerian origin: immunohistochemical analysis of a case and review of the literature. Int J Gynecol Cancer 2007; 17: 705–9. ArticlePubMed
- 5. Silverberg SG, Major FJ, Blessing JA, et al. Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A Gynecologic Oncology Group pathologic study of 203 cases. Int J Gynecol Pathol 1990; 9: 1–19. ArticlePubMed
- 6. Saglam O, Husain S, Toruner G. AKT, EGFR, c-ErbB-2, and c-kit expression in uterine carcinosarcoma. Int J Gynecol Pathol 2013; 32: 493–500. ArticlePubMed
- 7. Brinkmann D, Ryan A, Ayhan A, et al. A molecular genetic and statistical approach for the diagnosis of dual-site cancers. J Natl Cancer Inst 2004; 96: 1441–6. ArticlePubMed
- 8. Gallardo A, Matias-Guiu X, Lagarda H, et al. Malignant mullerian mixed tumor arising from ovarian serous carcinoma: a clinicopathologic and molecular study of two cases. Int J Gynecol Pathol 2002; 21: 268–72. ArticlePubMed
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- A successfully treated primary peritoneal carcinosarcoma and serous carcinoma of stage IIIC rescued from hypovolemic shock due to tumor rupture
Che-Cheng Huang, Horng-Jyh Tsai, Shih Hsuan Huang, Victor C. Kok
Taiwanese Journal of Obstetrics and Gynecology.2019; 58(2): 296. CrossRef - Ovarian Carcinosarcoma and Concurrent Serous Tubal Intraepithelial Carcinoma With Next-Generation Sequencing Suggesting an Origin From the Fallopian Tube
Sharlene Helene C. See, Amir Behdad, Kruti P. Maniar, Luis Z. Blanco
International Journal of Surgical Pathology.2019; 27(5): 574. CrossRef - Progression inference for somatic mutations in cancer
Leif E. Peterson, Tatiana Kovyrshina
Heliyon.2017; 3(4): e00277. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-

Peritoneal Carcinosarcoma and Ovarian Papillary Serous Carcinoma Are the Same Origin: Analysis of TP53 Mutation and Microsatellite Suggests a Monoclonal Origin



Fig. 1. The pericolic mass shows both carcinomatous and sarcomatous components composed of complex and interconnecting papillary structures lined by stratified malignant epithelial cells and a malignant cartilaginous element (A), but the ovarian mass contains only a carcinomatous component (B). On immunohistochemical staining, both the carcinomatous and the sarcomatous components in the pericolic mass (C) and the carcinomatous component of the ovarian tumor (D) show strong immunopositivity for p53.
Fig. 2. An identical somatic point mutation (TGT to TAT; p.C238Y) is detected in exon 7 of TP53 in both the carcinomatous and the sarcomatous component of the pericolic tumor mass (A) and in the epithelial component of the ovarian tumor (B).
Fig. 3. Several peaks show the same migration pattern, and each main peak of the five markers is seen at the same migration length in the pericolic tumor mass (A) and the epithelial component of the ovarian tumor (B).
Fig. 1.
Fig. 2.
Fig. 3.
Peritoneal Carcinosarcoma and Ovarian Papillary Serous Carcinoma Are the Same Origin: Analysis of TP53 Mutation and Microsatellite Suggests a Monoclonal Origin

