 E-submission
E-submission
Search
- Page Path
- HOME > Search
Review
- Diagnosis of interstitial lung diseases: from Averill A. Liebow to artificial intelligence
- Eunhee S. Yi, Paul Wawryko, Jay H. Ryu
- J Pathol Transl Med. 2024;58(1):1-11. Published online January 10, 2024
- DOI: https://doi.org/10.4132/jptm.2023.11.17
- 8,556 View
- 444 Download
- 2 Web of Science
- 2 Crossref
-
 Abstract
Abstract
 PDF
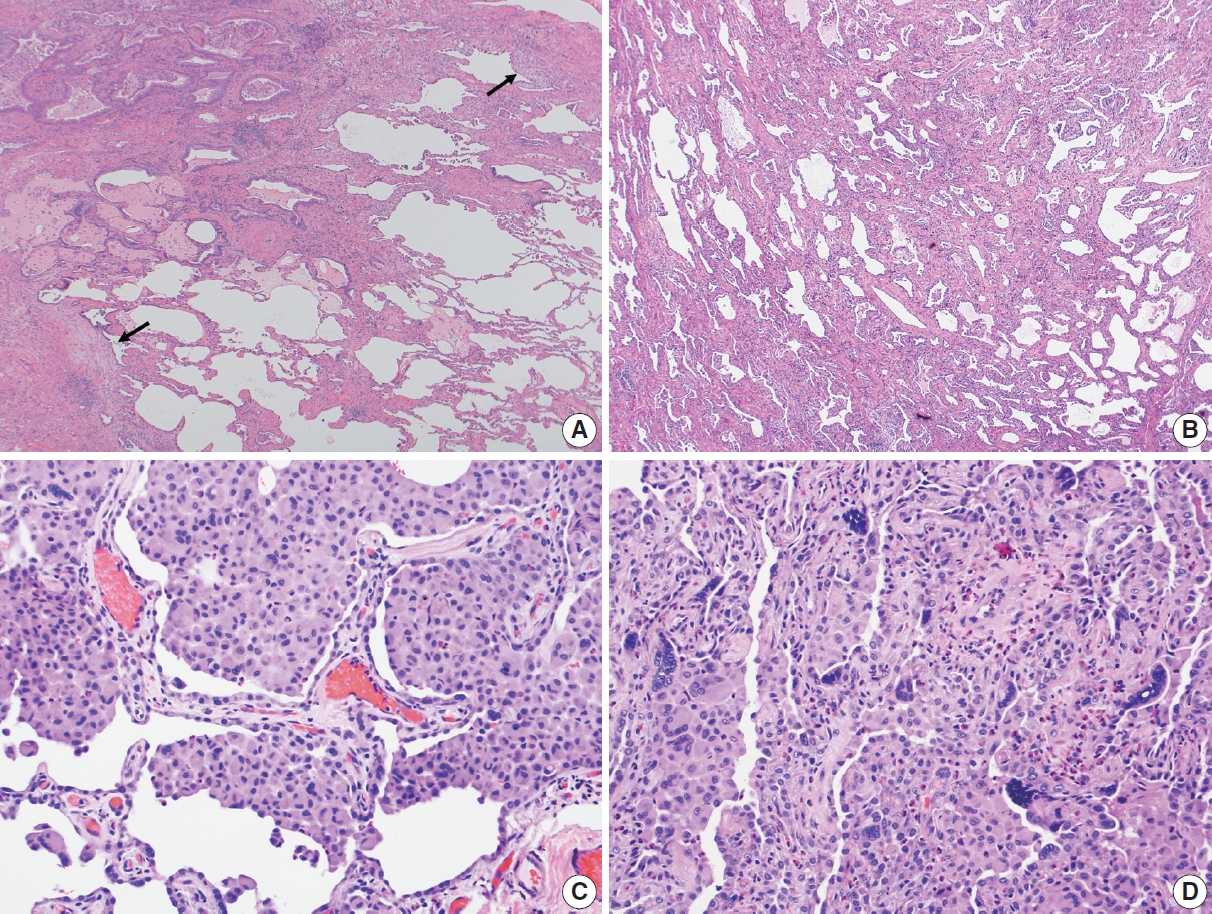
PDF - Histopathologic criteria of usual interstitial pneumonia (UIP)/idiopathic pulmonary fibrosis (IPF) were defined over the years and endorsed by leading organizations decades after Dr. Averill A. Liebow first coined the term UIP in the 1960s as a distinct pathologic pattern of fibrotic interstitial lung disease. Novel technology and recent research on interstitial lung diseases with genetic component shed light on molecular pathogenesis of UIP/IPF. Two antifibrotic agents introduced in the mid-2010s opened a new era of therapeutic approaches to UIP/IPF, albeit contentious issues regarding their efficacy, side effects, and costs. Recently, the concept of progressive pulmonary fibrosis was introduced to acknowledge additional types of progressive fibrosing interstitial lung diseases with the clinical and pathologic phenotypes comparable to those of UIP/IPF. Likewise, some authors have proposed a paradigm shift by considering UIP as a stand-alone diagnostic entity to encompass other fibrosing interstitial lung diseases that manifest a relentless progression as in IPF. These trends signal a pendulum moving toward the tendency of lumping diagnoses, which poses a risk of obscuring potentially important information crucial to both clinical and research purposes. Recent advances in whole slide imaging for digital pathology and artificial intelligence technology could offer an unprecedented opportunity to enhance histopathologic evaluation of interstitial lung diseases. However, current clinical practice trends of moving away from surgical lung biopsies in interstitial lung disease patients may become a limiting factor in this endeavor as it would be difficult to build a large histopathologic database with correlative clinical data required for artificial intelligence models.
-
Citations
Citations to this article as recorded by
- Identification of early genes in the pathophysiology of fibrotic interstitial lung disease in a new model of pulmonary fibrosis
Nathan Hennion, Corentin Bedart, Léonie Vandomber, Frédéric Gottrand, Sarah Humez, Cécile Chenivesse, Jean-Luc Desseyn, Valérie Gouyer
Cellular and Molecular Life Sciences.2025;[Epub] CrossRef - Radiological Insights into UIP Pattern: A Comparison Between IPF and Non-IPF Patients
Stefano Palmucci, Miriam Adorna, Angelica Rapisarda, Alessandro Libra, Sefora Fischetti, Gianluca Sambataro, Letizia Antonella Mauro, Emanuele David, Pietro Valerio Foti, Claudia Mattina, Corrado Spatola, Carlo Vancheri, Antonio Basile
Journal of Clinical Medicine.2025; 14(12): 4162. CrossRef
- Identification of early genes in the pathophysiology of fibrotic interstitial lung disease in a new model of pulmonary fibrosis
Case Report
- Acute Interstitial Pneumonia (Hamman-Rich Syndrome): An Autopsy Case.
- Han Kyeom Kim, Ae Ree Kim, Min Ji Jeoung, Won Hee Seo, Jee yeoun Lee, Su Hyun Park
- Korean J Pathol. 1997;31(4):366-374.
- 2,744 View
- 35 Download
-
Abstract
PDF
- Acute interstitial pneumonia is a fulminant disease of unknown etiology that usually occurs in a previously healthy person and produces the histologic findings of the organizing phase of diffuse alveolar damage. We experienced an autopsy case of acute interstitial pneumonia of unknown etiology. The patient was a 48 year old man who had been healthy and had not been exposed to organic dusts or other toxic materials. The chief complaints represented were dyspnea and a dry cough for several weeks before hospitalization, and the chest radiographs showed bilateral interstitial infiltrates. Patchy consolidation of air space was also identified and ground-glass attenuation similar to those described in ARDS was detected on high-resolution computed tomography. Steroid pulse therapy, mechanical ventilation, and antibiotics for superimposed bacterial infection were performed, but the symptoms did not improve and the patient died of generalized respiratory insufficiency and severe hypoxemia 2 1/2 months after hospitalization. At autopsy the macroscopic and microscopic findings were confined mainly to the lungs. On the whole, both lungs were firm in consistency and the external surface showed a cobblestone appearance. The cut surface showed almost complete replacement of the normal lung parenchyma with gray to yellow fibrous tissue with a little residual functional area remaining. The pathology of both open lung biopsy and autopsy tissue showed marked hyperplasia of type II pneumocytes, hyaline membrane formation, thickening of the alveolar wall due to extensive fibroblast proliferation, and relatively abundant young collagen deposition in the interstitium. An immunohistochemical stain for cytokeratin revealed epithelial hyperplasia and showed that the alveolar spaces were markedly shrunken by fibrous tissue.
Original Article
- Pathology of Chronic Interstitial Lung Disease.
- Dong Hwan Shin
- Korean J Pathol. 1998;32(1):1-8.
- 2,969 View
- 52 Download
-
Abstract
PDF
- Interstitial lung disease is a generic term for a heterogeneous group of lung disease that primarily affect the interstitium although the disease is not clearly restricted to the interstitium. The majority of interstitial lung diseases represent inflammatory insults to the microscopic anatomic space bounded by the basement membrane of epithelial and endothelial cells, which may occur as slowly developing process and ultimately end up as end-stage honeycomb fibrosis. The currently prevalent classification of interstitial pneumonia with practical utility and easy reproducibility pertaining only to idopathic interstitial pneumonia encompasses several different entities some of which may represent different aspects of the same condition. Honeycomb fibrosis is usually caused by a variety of pulmonary disease including chronic interstitial lung disease. It is important to recognize that usual inter-stitial pneumonia and honeycomb fibrosis are not synonymous. In the era of chemotherapy for malignant tumor, aggressive immunosuppression for autoimmune diseases and transplant recipients and acquired immunodeficiency syndrome, lung disease in the immunocompromised host has been common. Diagnostic lung biopsy becomes increasingly needed because proper treatment of interstitial lung disease relies on correct morphologic diagnosis. This review summarizes the pathologic spectrum of idiopathic interstitial pneumonias together with other inflammatory process with known or suggestive etiologies simulating interstitial pneumonias.
 First
First Prev
Prev


