Cutaneous soft tissue tumors in the 5th edition of the World Health Organization classification of skin tumors: key updates and new entities
Article information

Abstract
The 5th edition of the World Health Organization (WHO) classification of skin tumors introduces a dedicated chapter on cutaneous soft tissue tumors, providing a comprehensive, standardized reference with updated diagnostic criteria that directly inform routine dermatopathology practice and molecular diagnostics. This edition incorporates several key changes, including newly recognized entities such as EWSR1::SMAD3-rearranged fibroblastic tumor, neurotrophic tyrosine receptor kinase (NTRK)–rearranged spindle cell neoplasm, superficial CD34-positive fibroblastic tumor, and CRTC1::TRIM11 cutaneous tumor. Diagnostic terminology has also been refined; for example, the term ‘atypical intradermal smooth muscle neoplasm’ replaces ‘cutaneous leiomyosarcoma’ for lesions confined to the dermis, whereas the designation leiomyosarcoma is reserved for tumors with overt subcutaneous infiltration. In addition, epithelioid fibrous histiocytoma has been reassigned to the family of tumors of uncertain differentiation. This review summarizes the key updates and newly recognized entities in the chapter on cutaneous soft tissue tumors in the 5th edition of the WHO classification of skin tumors, emphasizing their clinicopathological and molecular implications.
INTRODUCTION
The 5th edition of the World Health Organization (WHO) classification of skin tumors, volume 12 of the WHO classification of tumors series, was published online in 2023 and in print in September 2025 [1]. This series is widely regarded as the gold standard for tumor diagnosis, providing an authoritative synthesis of clinical, histopathological, and molecular criteria, as well as practical diagnostic approaches. Since the publication of the 4th edition of the WHO classification of skin tumors in 2018 [2], significant advances have been made in soft tissue pathology, particularly in identifying novel molecular and genetic alterations, discovering new immunohistochemical markers, and clarifying underlying biological mechanisms [3,4]. The 5th edition incorporates these advances, thereby reflecting major progress in the understanding of soft tissue tumors.
The dedicated chapter on soft tissue tumors in the 5th edition of the WHO classification of skin tumors includes entities that are confined to the skin or characterized by distinctive cutaneous features. This chapter maintains the organizational structure of the previous edition, classifying soft tissue tumors into seven main families: (1) adipocytic tumors, (2) fibroblastic, myofibroblastic, and fibrohistiocytic tumors, (3) vascular tumors, (4) pericytic and perivascular tumors, (5) smooth muscle tumors, (6) neural tumors, and (7) tumors of uncertain differentiation (Table 1).
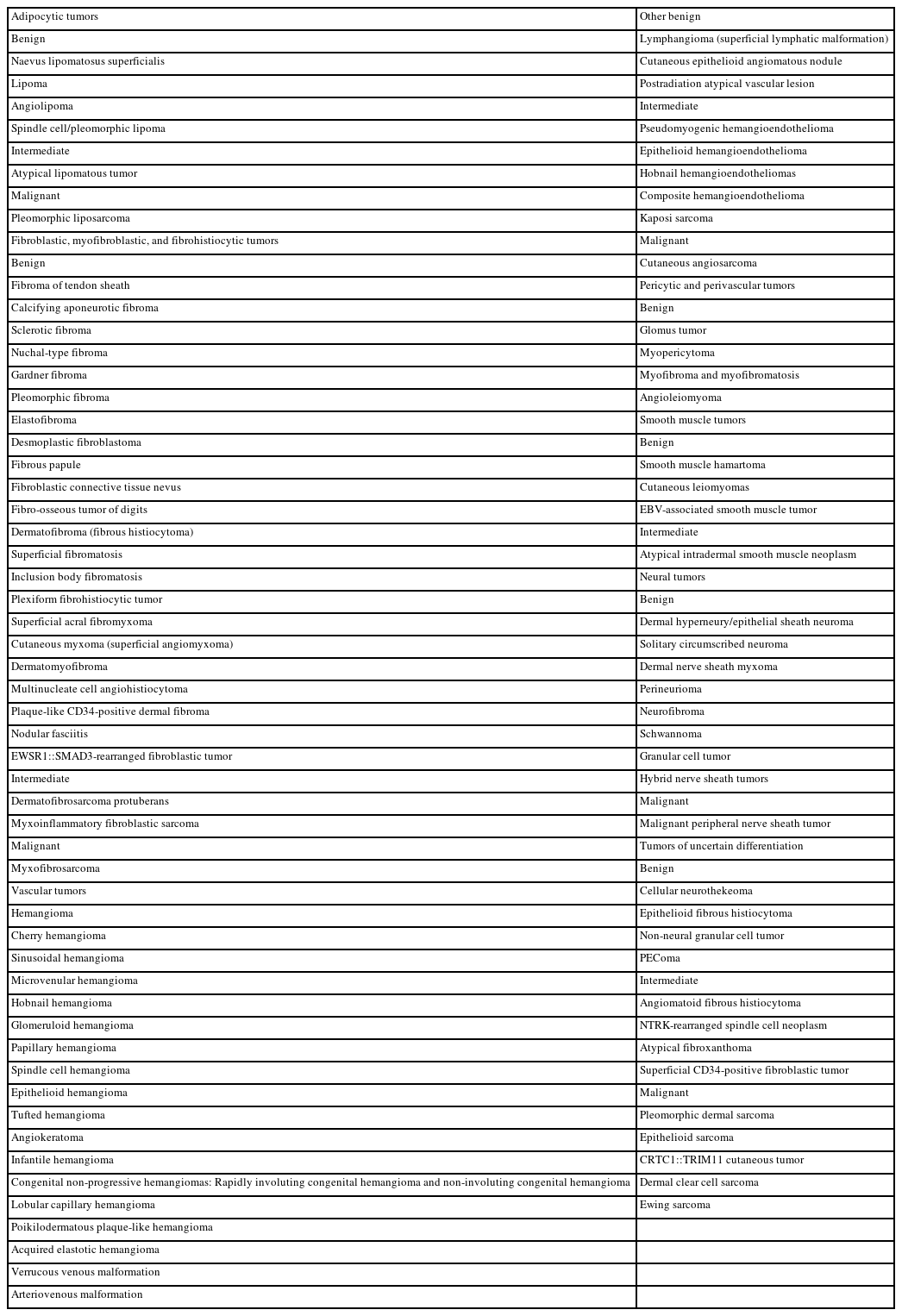
Soft tissue tumors that are included in the 5th edition of the WHO classification of skin tumors
Cutaneous soft tissue tumors present considerable diagnostic challenges due to their diversity and overlapping histological features. The 5th edition recognizes several newly defined entities that reflect recent advances in tumor biology, molecular genetics, and diagnostic approaches [5,6]. A thorough understanding of these updates is essential for accurate diagnosis, optimal patient management, and improved prognostication.
This review summarizes key updates to cutaneous soft tissue tumors in the 5th edition of the WHO classification, highlighting newly recognized entities and tumors incorporated from other WHO volumes, along with their clinical, histological, and molecular features, and recent advances in tumor pathogenesis and immunohistochemistry (IHC). It is intended to serve as a practical reference for practicing pathologists, with particular emphasis on updated diagnostic criteria and newly introduced molecular correlates.
The updates in the 5th edition of the WHO classification of skin tumors are summarized as follows: newly recognized tumor entities and tumors incorporated from other WHO volumes (Table 2); newly identified molecular genetic alterations and immunohistochemical markers (Table 3); tumors with revised terminology (Table 4); and tumors reassigned to different families (Table 5).
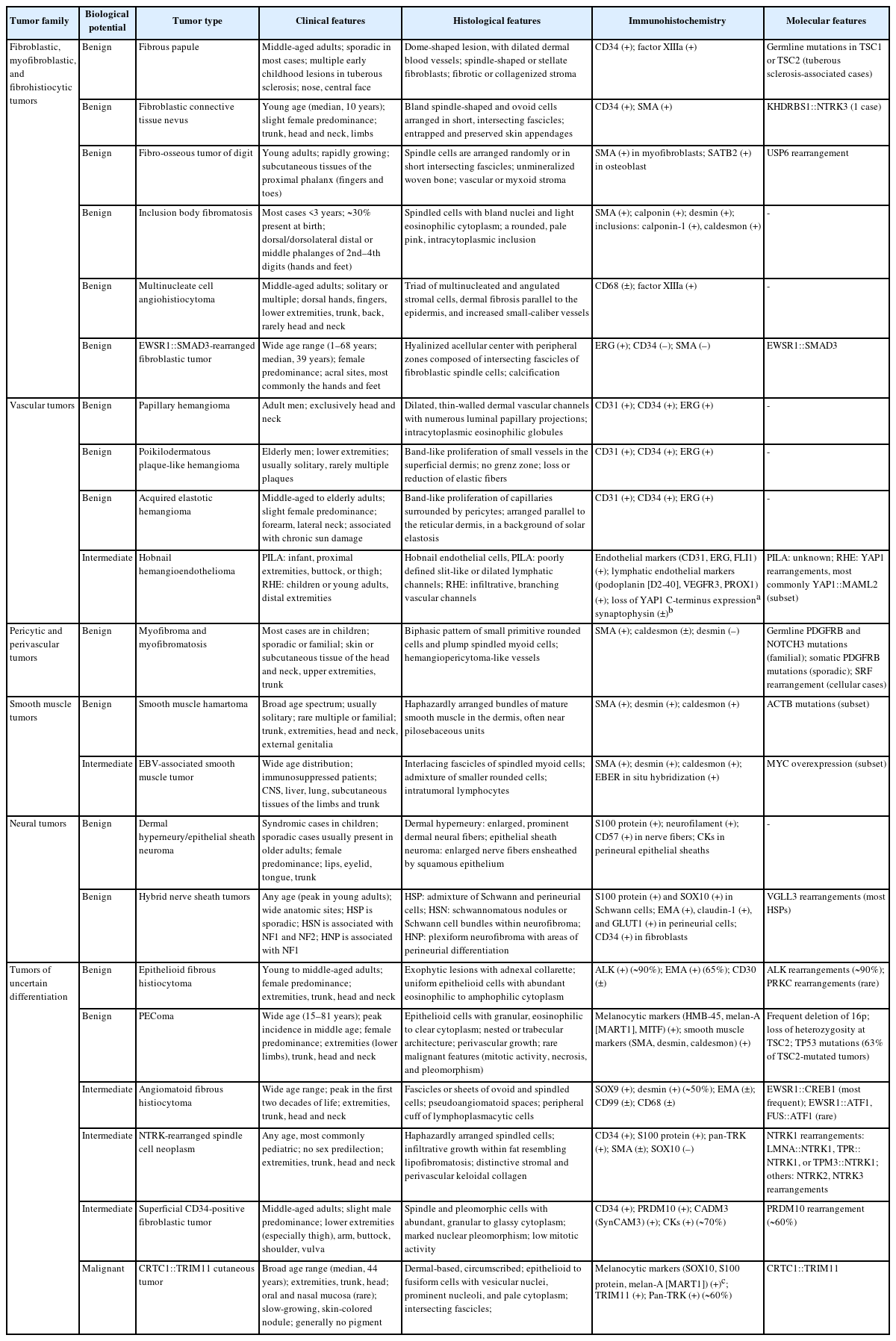
Newly recognized tumor entities and selected tumors newly incorporated from other volumes in the 5th edition of the WHO classification of skin tumors
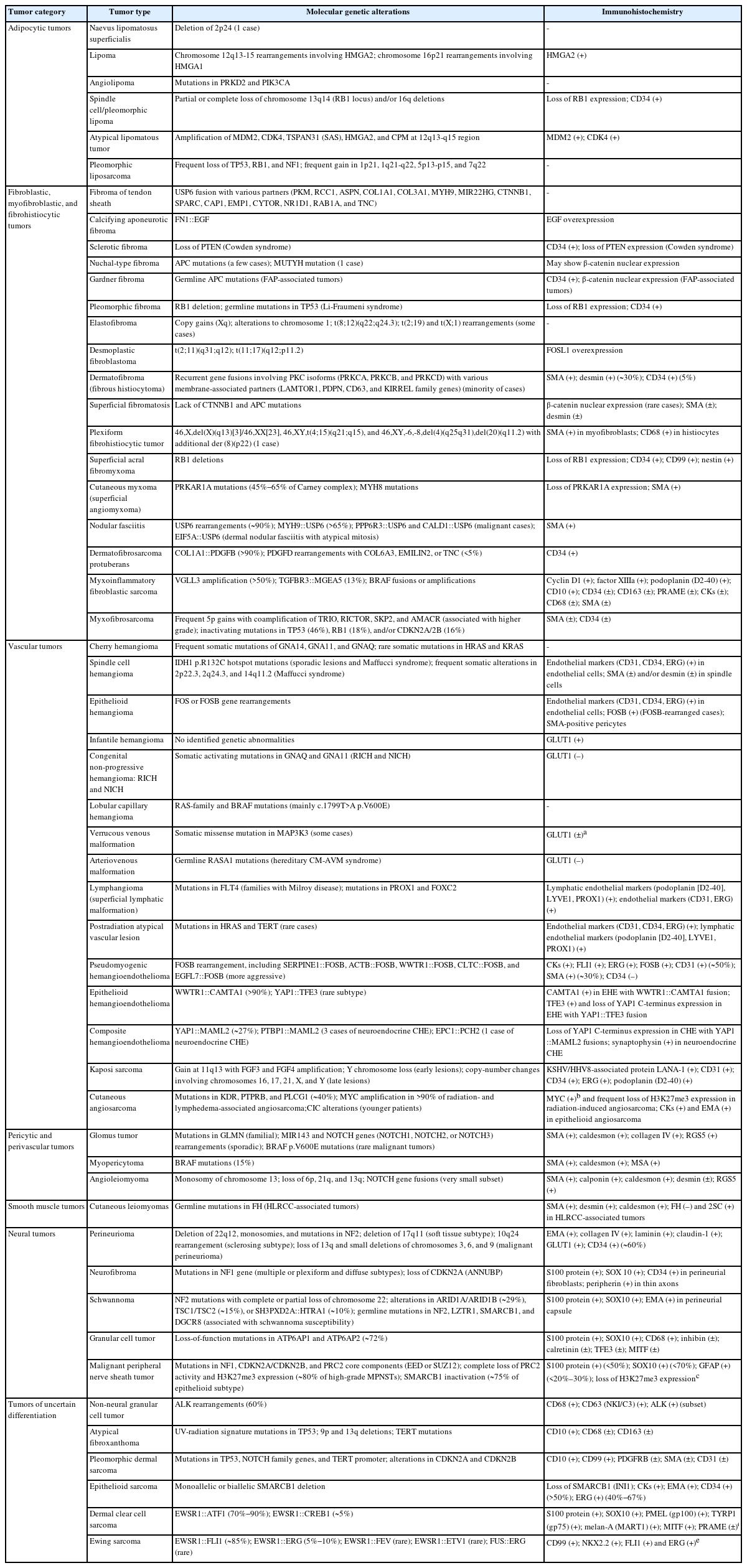
Newly identified molecular genetic alterations and immunohistochemical markers in selected cutaneous soft tissue tumors
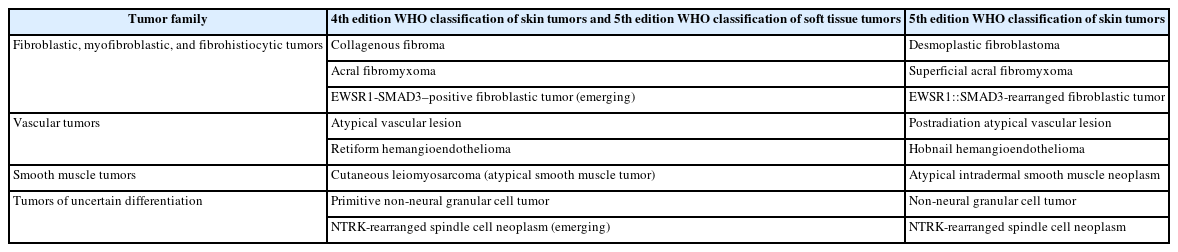
Soft tissue tumors with terminology changes in the 5th WHO classification of skin tumors
Selected soft tissue tumors whose tumor family has been changed in the 5th edition of the WHO classification of skin tumors
ADIPOCYTIC TUMORS
Key updates
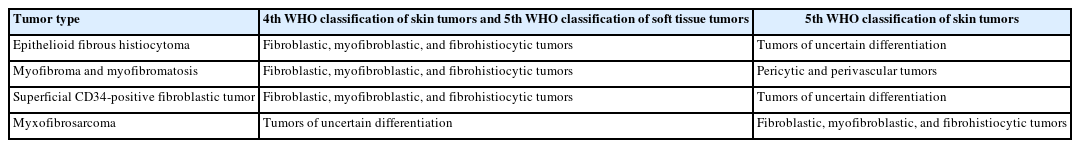
In the 5th WHO classification, nevus lipomatosus superficialis is defined as a benign cutaneous hamartoma characterized by dermal ectopic adipose tissue rather than as a developmental anomaly [7]. It is classified into two subtypes: classic and solitary. Additionally, among conventional lipomas, osteolipoma and chondrolipoma are recognized as distinct subtypes, defined by osseous and cartilaginous metaplasia, respectively [8]. In contrast, previously described subtypes, including fibrolipoma, myxolipoma, angiomyxolipoma, sclerotic lipoma, and chondroid lipoma, have been removed from the current WHO classification [9]. Cellular angiolipoma is defined as a subtype with a predominant vascular component and a sparse adipocytic component [10,11]. Spindle cell lipoma (SCL) and pleomorphic lipoma (PL) represent a morphological spectrum of the same benign adipocytic neoplasm [12], sharing RB1 deletion at chromosome 13q14, supporting their close biological relationship. Recognized morphologic patterns include fat-poor, myxoid, and pseudoangiomatous variants of SCL/PL (Fig. 1A, B).
Dermal myxoid spindle cell lipoma. (A) An unencapsulated, infiltrative tumor is present in the dermis. (B) The tumor is composed of bland, short spindle cells, mature adipocytes, and ropy collagen bundles in a prominent myxoid stroma.
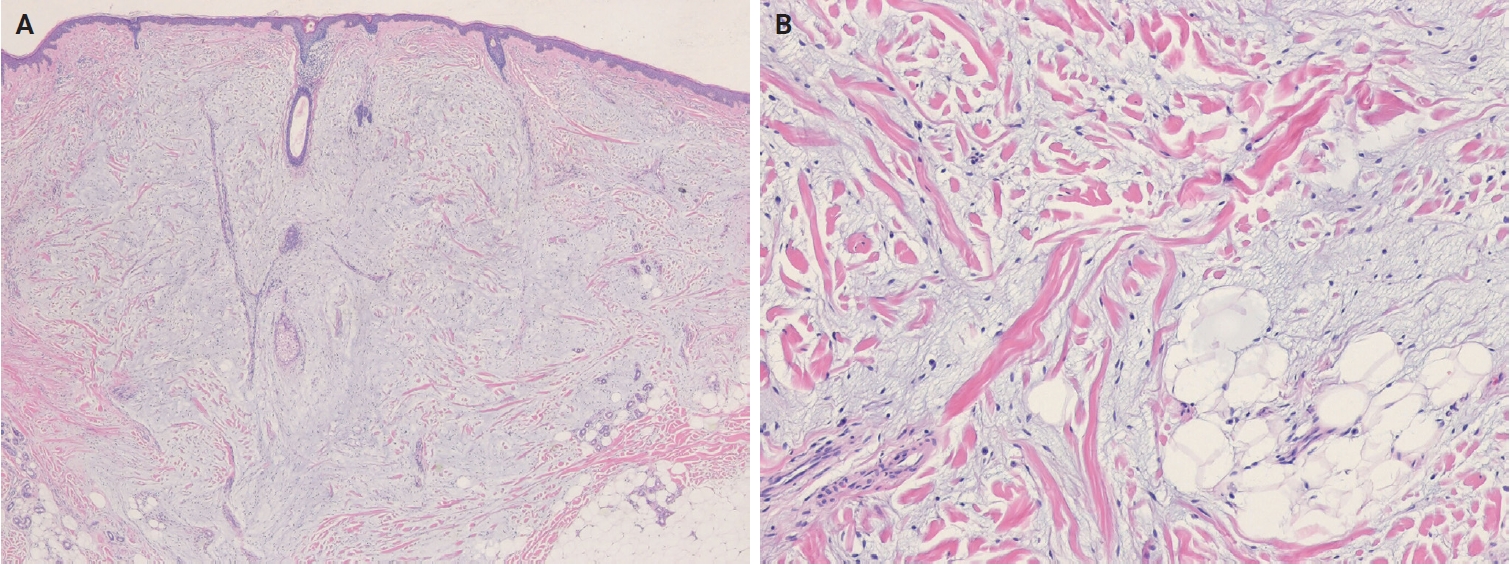
Atypical lipomatous tumor (ALT) is defined as an adipocytic neoplasm of borderline malignancy characterized by focal nuclear atypia and MDM2 and/or CDK4 amplification [13]. Primary cutaneous or subcutaneous ALTs are exceedingly rare [14,15]. Mature heterologous elements, such as osseous or myogenic tissue, may occasionally be present; this does not indicate dedifferentiation [16,17]. The terms ALT and well-differentiated liposarcoma (WDLPS) are synonymous, as they describe morphologically and genetically identical neoplasms. However, the term ‘WDLPS’ should be avoided for superficial tumors because these lesions typically exhibit less aggressive behavior. Pleomorphic liposarcoma (PLPS) is a high-grade, pleomorphic spindle cell sarcoma containing variable numbers of pleomorphic lipoblasts without components of WDLPS or other differentiation (Fig. 2A, B) [18]. An epithelioid subtype of PLPS, characterized by epithelioid cytomorphology, has been recognized as a distinct subtype [19]. The absence of MDM2 amplification is a key diagnostic criterion distinguishing PLPS from ALT and dedifferentiated liposarcoma.
Dermal pleomorphic liposarcoma. (A) The tumor is composed of an undifferentiated spindle cell sarcoma containing numerous pleomorphic lipoblasts. (B) Pleomorphic lipoblasts have lipid droplets indenting the pleomorphic, hyperchromatic nuclei. Signet-ring lipoblasts are also present. Reprinted from Patel and Folpe. Pleomorphic liposarcoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. [18], with permission from IARC Press.
FIBROBLASTIC, MYOFIBROBLASTIC, AND FIBROHISTIOCYTIC TUMORS
Key updates
The fibroblastic, myofibroblastic, and fibrohistiocytic tumor family has been expanded in the 5th WHO classification to include several newly described entities, such as fibrous papule, fibroblastic connective tissue nevus (FCTN), fibro-osseous tumor of digits (FOTD), inclusion body fibromatosis (IBF), multinucleate cell angiohistiocytoma (MCA), and EWSR1::SMAD3-rearranged fibroblastic tumor (ESRFT). Epithelioid fibrous histiocytoma (EFH), previously classified as a subtype of dermatofibroma (fibrous histiocytoma), is now recognized as a distinct entity characterized by anaplastic lymphoma kinase (ALK) overexpression due to ALK rearrangement and reassigned to the family of tumors of uncertain differentiation. Myxofibrosarcoma, previously placed in the family of tumors of uncertain differentiation, has been reclassified within the fibroblastic, myofibroblastic, and fibrohistiocytic tumor family. The fibroblastic tumor previously designated as EWSR1::SMAD3-positive (emerging) is now formally recognized as an ESRFT.
Additionally, the 5th WHO classification identifies recurrent USP6 gene rearrangements with multiple fusion partners, including PKM, RCC1, ASPN, COL1A1, COL3A1, MYH9, MIR22HG, CTNNB1, SPARC, CAP1, EMP1, CYTOR, NR1D1, RAB1A, and TNC, in fibroma of tendon sheath [20,21], supporting the concept that some cases represent a tenosynovial variant of nodular fasciitis [22]. Calcifying aponeurotic fibroma (CAF) harbors a recurrent FN1::EGF fusion, leading to aberrant epidermal growth factor (EGF) expression [23]. This fusion has also been identified in a subset of lipofibromatosis-like tumors, which may represent CAF variants lacking calcification [24]. Patients younger than 5 years are at higher risk of local recurrence [25]. In sclerotic fibroma, loss of phosphatase and tensin homolog (PTEN) expression on IHC may suggest Cowden syndrome [26]. Multiple pleomorphic fibromas in young patients may indicate an underlying genetic cancer syndrome, such as Li-Fraumeni syndrome associated with TP53 germline mutations [27].
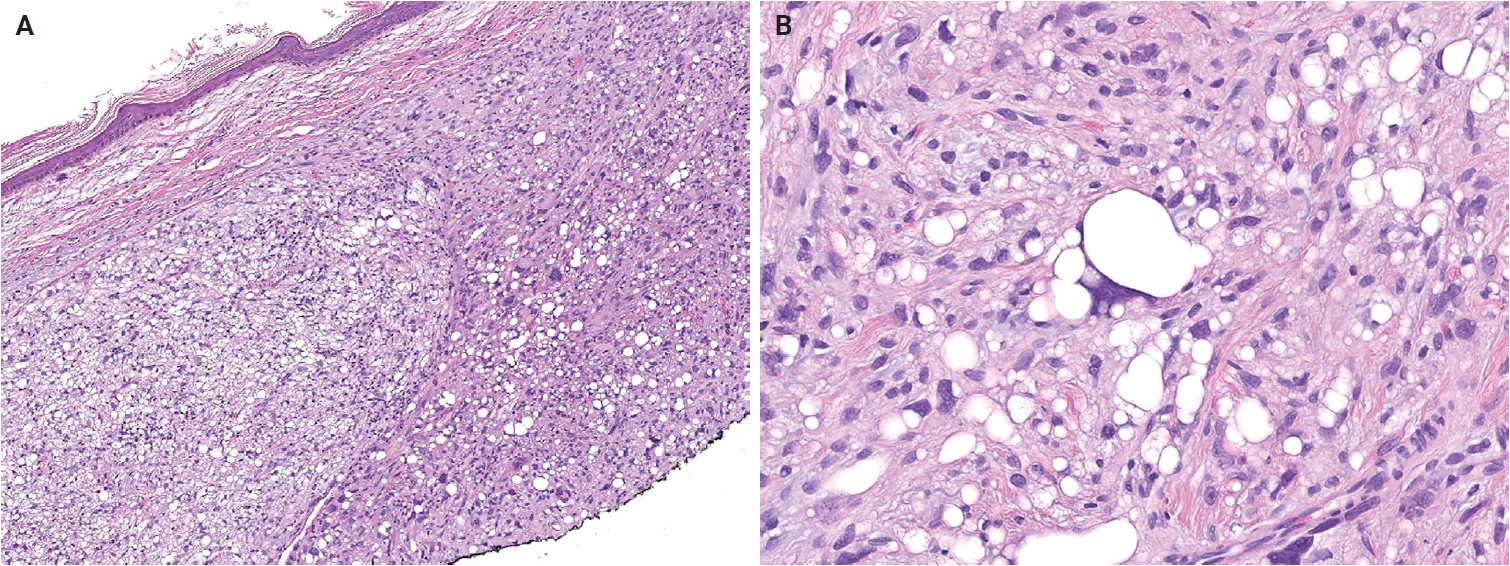
In the 5th edition, dermatofibroma (fibrous histiocytoma) is formally classified into five subtypes—classic, cellular, aneurysmal, atypical, and deep fibrous histiocytoma—each with distinct clinical and pathological features (Fig. 3A, B) [28]. Recurrent gene fusions involving PKC isoforms (PRKCA, PRKCB, and PRKCD) and various genes encoding membrane-associated proteins (LAMTOR1, PDPN, CD63, and KIRREL family genes) have been identified in a subset of these tumors [29-31]. Superficial fibromatosis is defined as a nodular, locally recurrent fibroblastic/myofibroblastic proliferation that includes palmar, plantar, and penile subtypes [32]. Although superficial fibromatosis involves activation of the WNT/β-catenin signaling pathway, it lacks the CTNNB1 and APC mutations characteristic of desmoid fibromatosis [33]. Plexiform fibrohistiocytic tumor exhibits diverse cytogenetic alterations, including 46,X,del(X)(q13)[3]/46,XX[23]; 46,XY,t(4;15)(q21;q15); and 46,XY,-6,-8,del(4)(q25q31),del(20)(q11.2) with additional der(8)(p22) [34-36].
Cellular dermatofibroma (fibrous histiocytoma). (A) Scanning magnification shows a dermal tumor with lateral borders interdigitating with the adjacent dermis. The overlying epidermis shows acanthosis. (B) The tumor is highly cellular and composed of spindle cells arranged in a storiform pattern. Hyalinized collagen fibers are present.
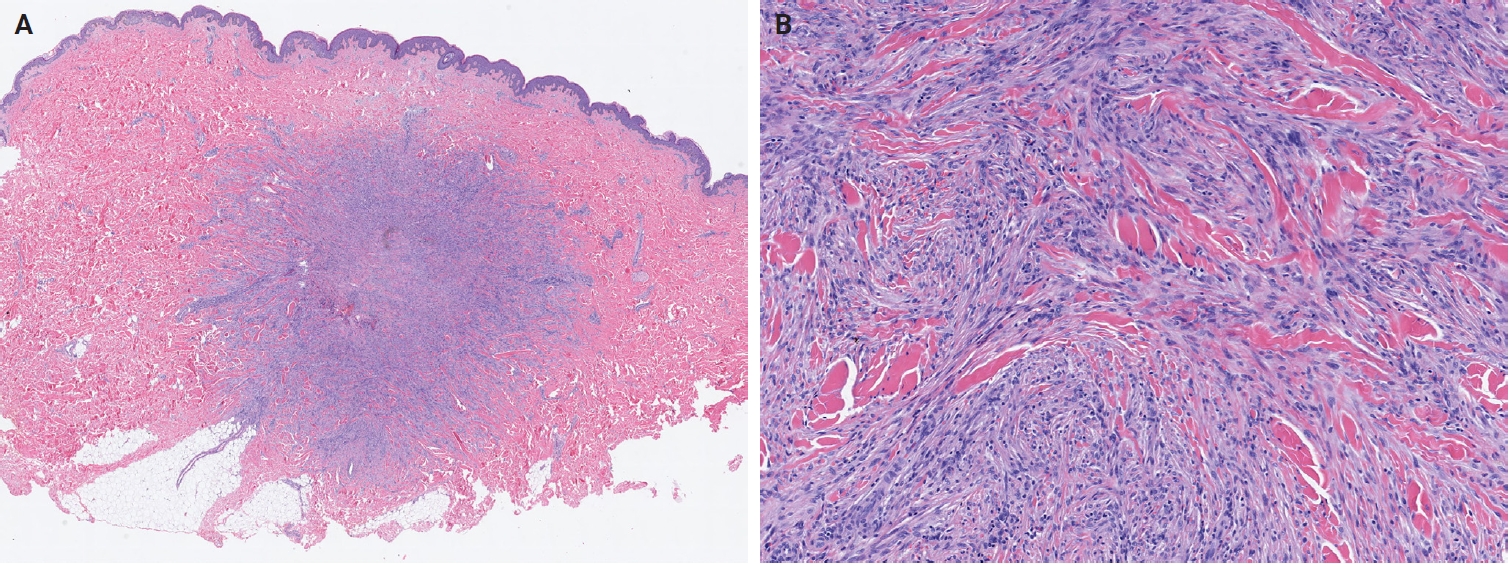
In cutaneous myxoma (superficial angiomyxoma), approximately 45%–65% of Carney complex-associated cases harbor PRKAR1A mutations [37,38], while alternative genetic pathways, including MYH8 mutations, have also been identified [39]. Nodular fasciitis shows USP6 rearrangements in approximately 90% of cases, with MYH9::USP6 accounting for >65% of these [40]. Atypical or malignant nodular fasciitis cases harboring PPP6R3::USP6 or CALD1::USP6 fusions have rarely been reported [41,42]. An EIF5A::USP6 fusion has also been identified in dermal nodular fasciitis of the forearm with atypical mitoses [43]. In dermatofibrosarcoma protuberans (DFSP), COL1A1::PDGFB remains the hallmark translocation, while alternative PDGFD rearrangements involving COL6A3, EMILIN2, or TNC have also been described [44-46]. Genomic gains of COL1A1::PDGFB and alterations in the mechanistic target of rapamycin (mTOR) pathway have been associated with fibrosarcomatous transformation [46,47]. The subtypes of DFSP have been standardized to include classic DFSP, giant cell fibroblastoma, myxoid DFSP, pigmented DFSP, myoid DFSP, plaque-like DFSP, DFSP with fibrosarcomatous transformation, and other morphological patterns (granular cell, sclerosing, and atrophic types) (Fig. 4A, B) [48].
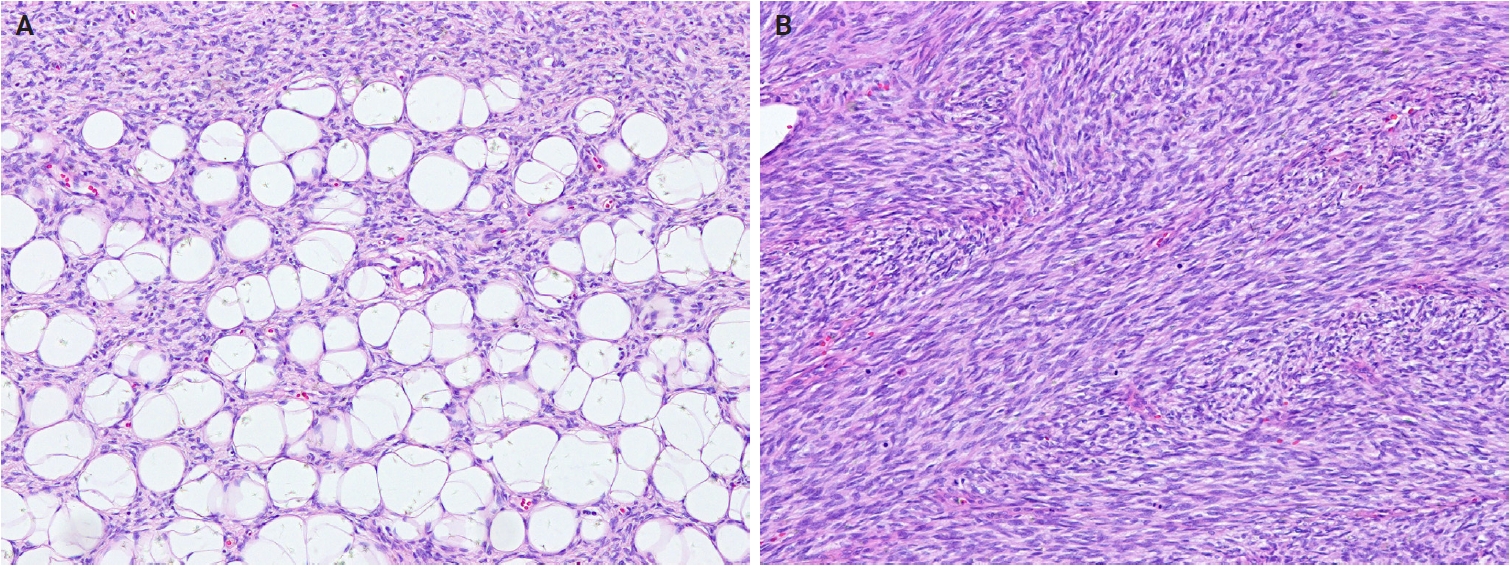
Dermatofibrosarcoma protuberans with fibrosarcomatous transformation. (A) Uniform spindle-shaped tumor cells infiltrate the subcutaneous fat in a honeycomb pattern, consistent with conventional dermatofibrosarcoma protuberans. (B) Areas of fibrosarcomatous transformation show increased cellularity, a fascicular growth pattern, and frequent mitotic figures.
In myxoinflammatory fibroblastic sarcoma (MIFS), VGLL3 amplification at 3p11-p12 occurs in >50% of cases, resulting in a supernumerary ring chromosome [49]. Histologically, MIFS is characterized by atypical fibroblastic cells with macronucleoli and a mixed inflammatory infiltrate in variably myxoid and hyalinized stroma (Fig. 5A, B) [50]. A t(1;10) translocation involving MGEA5 (OAG) (10q24) and TGFBR3 (1p22) is present in approximately 13% of cases and has also been identified in pure hemosiderotic fibrolipomatous tumors (HFLTs), hybrid MIFS-HFLT lesions, and pleomorphic hyalinizing angiectatic tumors [51-53]. BRAF fusions or amplifications have also been identified [54]. Myxofibrosarcomas exhibit frequent chromosomal rearrangements and copy-number alterations, even in low-grade tumors, with increasing genomic complexity in recurrent tumors showing histological progression [55-58]. Myxofibrosarcomas are characterized by somatic copy-number alterations, with several oncogenes—TRIO, RICTOR, SKP2, and AMACR—located on 5p, which is frequently gained, often co-amplified, and associated with higher histological grade [59-61]. Alterations in tumor suppressor genes, including TP53 (46%), RB1 (18%), and CDKN2A/CDKN2B (16%), occur more frequently than oncogene amplifications involving CDK6, CCND1, or MDM2 [61].
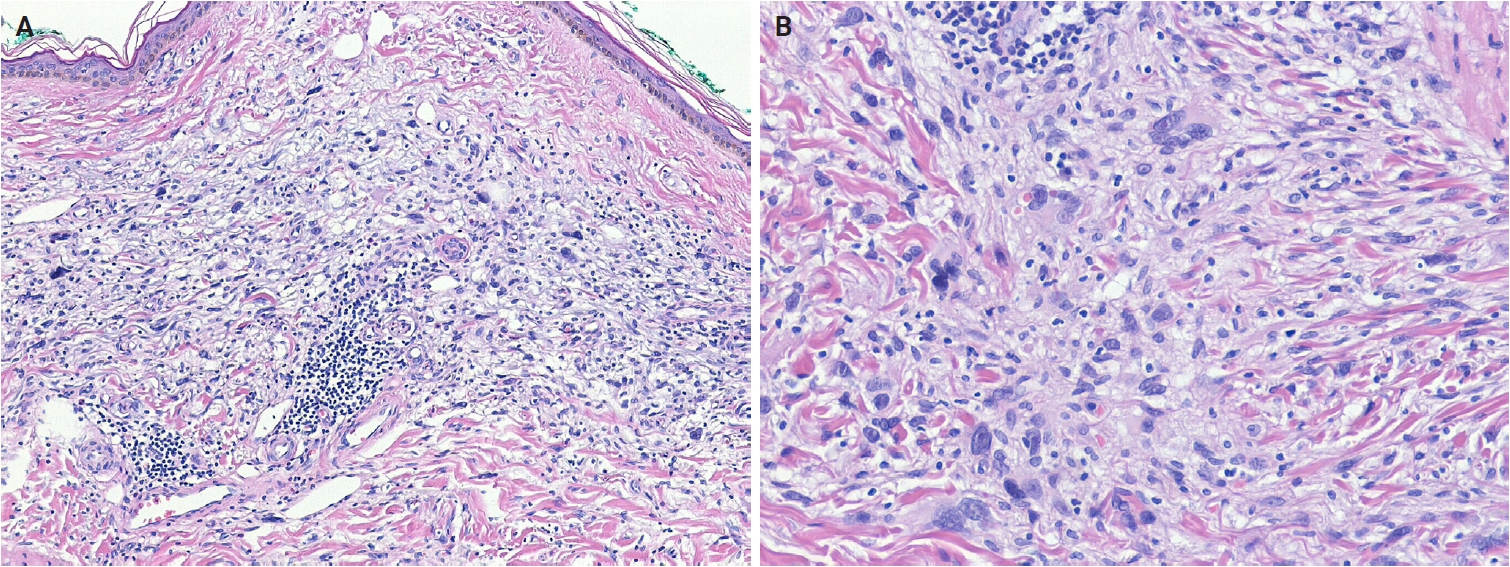
Myxoinflammatory fibroblastic sarcoma. (A) The tumor shows an ill-defined dermal lesion composed of pleomorphic tumor cells admixed with inflammatory cells in a myxoid stroma. (B) Pleomorphic multinucleated giant cells with prominent nucleoli are present.
Fibrous papule
Fibrous papule is a benign lesion of presumed fibroblastic origin that most commonly arises on the nose or central face [62]. It predominantly affects middle-aged adults [63]. Clinically, it presents as a solitary, sporadic, dome-shaped, sessile papule. The presence of multiple (syndromic) fibrous papules is associated with tuberous sclerosis and, less commonly, with Birt-Hogg-Dubé syndrome or multiple endocrine neoplasia type 1 [64,65]. In tuberous sclerosis, multiple fibrous papules are associated with germline mutations in TSC1 or TSC2, leading to activation of the mTOR pathway. A similar mTOR-related mechanism has also been proposed for sporadic fibrous papules [66].
Fibrous papules are well-circumscribed papular lesions with a tan to white cut surface. Histologically, they are dome-shaped and composed of dilated dermal blood vessels, surrounded by fibrotic or collagenized stroma containing spindle-shaped or stellate fibroblasts [67]. The overlying epidermis may exhibit melanocytic hyperplasia [68]. Histological subtypes include hypercellular, pigmented, pleomorphic, clear cell, granular cell, epithelioid, and inflammatory variants.
Fibroblastic connective tissue nevus
FCTN is a benign, dermal mesenchymal lesion of the fibroblastic/myofibroblastic lineage, characterized by short, intersecting fascicles of bland spindle cells [69]. Lesions commonly occur on the trunk, head and neck, or extremities [70-72]. FCTN shows a slight female predominance and typically affects children and young individuals (median age, 10 years). Congenital cases have been reported, often presenting as large, infiltrated plaques on the trunk [73,74]. Clinically, FCTN usually manifests as a solitary, slowly growing, painless plaque or nodule, although rare presentations include agminate lesions [75,76], atrophic plaques [77], or exophytic tumors. A single case harboring the KHDRBS1::NTRK3 gene fusion has been reported [73].
Histologically, FCTN is a poorly circumscribed, dermal-based lesion localized primarily in the reticular dermis and often extending into the superficial subcutis. It comprises bland, spindle-shaped to ovoid cells arranged in short, intersecting fascicles, with preserved adnexal structures. Cellularity varies, mitotic figures are rare or absent, and a storiform pattern may occasionally be observed. Epidermal papillomatosis and ectopic dermal adipose tissue are frequently present [70]. Elastic fibers often decrease. Immunohistochemically, tumor cells express CD34 and smooth muscle actin (SMA), supporting fibroblastic/myofibroblastic differentiation [71].
Fibro-osseous tumor of digits
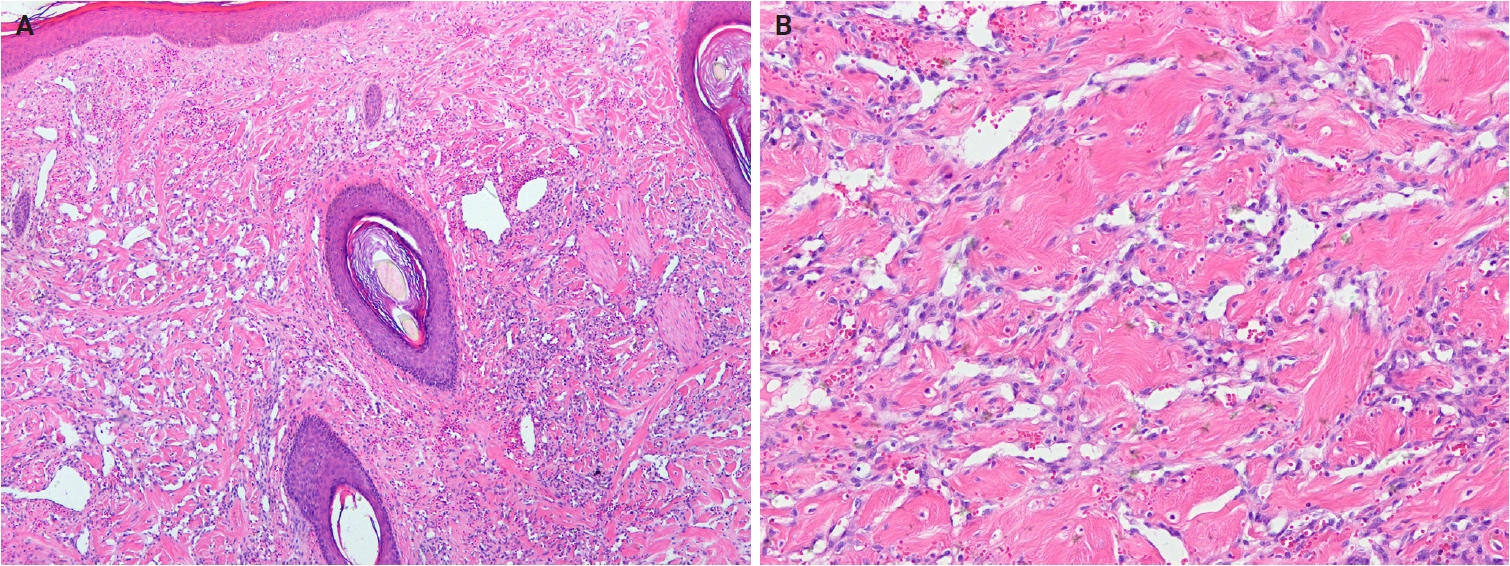
FOTD is a localized, self-limiting benign neoplasm that arises in the soft tissue of the digits [78]. It most commonly occurs around the proximal phalanx of the fingers and, less frequently, the toes [79-81]. FOTD occurs over a wide age range, with a predilection for young adults [79,80]. Clinically, it presents as a rapidly growing mass that may be painful and demonstrates irregular calcifications on imaging. Over time, the lesion hardens and becomes well-circumscribed [82]. Molecular studies have identified USP6 fusion genes in FOTD [80,83], a feature shared with nodular fasciitis, which shows overlapping morphology.
Histologically, FOTD shows nodular fasciitis-like areas with variable stages of bone formation. The lesion is hypercellular and composed of randomly arranged or short, intersecting fascicles of spindle cells with plump, fusiform nuclei containing fine chromatin, conspicuous nucleoli, and moderate amounts of eosinophilic cytoplasm. Mitoses are frequent but typical. The stroma is richly vascular and variably myxoid, containing fibrin, extravasated erythrocytes, scattered lymphocytes, and osteoclast-like giant cells. Spindle cells gradually blend into ill-defined trabeculae and sheets of unmineralized woven bone, which are rimmed by prominent osteoblasts with amphophilic cytoplasm and contain large osteocytes [82,84].
Inclusion body fibromatosis
IBF is a benign myofibroblastic tumor characterized by eosinophilic intracytoplasmic inclusions and a propensity for local recurrence [85]. It is subclassified into classic (digital) and non-classic (extradigital) subtypes. Classic IBF accounts for approximately 2% of pediatric fibroblastic tumors [86], with >80% of cases diagnosed before the age of three and approximately 30% present at birth [87]. There is no sex predilection. Rare adult cases have been reported [88]. Classic IBFs typically arise on the dorsal or dorsolateral aspects of the distal or middle phalanges of the second to fourth digits, less frequently the fifth digit, and usually spare the first digit [89]. Non-classic IBF involves the extremities, tongue, and breast [90]. Clinically, classic IBFs present as asymptomatic, dome-shaped or polypoid nodules, typically <20 mm in diameter [89]. They may occur synchronously or metachronously, occasionally involving multiple digits [91]; concurrent finger and toe involvement is rare. In contrast, non-classic IBFs usually present as a solitary mass or nodule.
Histologically, IBF comprises spindle cells with lightly eosinophilic cytoplasm and elongated, bland nuclei, exhibiting low mitotic activity. These cells form whorls, short interlacing fascicles, or storiform patterns within the collagenous dermis, often oriented perpendicularly to the epidermis and encircling adnexal structures, with occasional extension into deeper tissues [91]. Rounded, pale pink, intracytoplasmic inclusions (1.5–24 μm) are highlighted by trichrome (red), phosphotungstic acid–hematoxylin (dark purple), and Movat’s stains (pink) [89]. Immunohistochemically, tumor cells express vimentin, muscle actins (displaying peripheral ‘tram-track’ pattern), calponin, desmin, and CD99 [92]. The inclusions are positive for calponin-1 [93] and occasionally for caldesmon [89].
Multinucleate cell angiohistiocytoma
MCA is a benign, acquired lesion of fibrohistiocytic and vascular lineage [94]. It most commonly occurs on the dorsal surfaces of the hands, fingers, lower extremities, trunk, and back [95], with head and neck involvement being uncommon. Clinically, MCA typically presents in middle-aged adults as solitary or multiple smooth, erythematous papules or nodules. Some studies have reported a female predominance [96], whereas others have observed no significant sex difference [97]. The pathogenesis remains uncertain; proposed mechanisms include fibroblast proliferation, estrogen receptor α overexpression, and mast cell involvement [98,99].
Histologically, MCA exhibits a characteristic triad: multinucleated stromal cells, parallel stromal fibrosis, and proliferation of small-caliber vessels. Multinucleated stromal cells are usually angulated or stellate, with elongated cytoplasmic processes, and are often sparse, detectable only in deeper sections [97]. Stromal fibrosis runs parallel to the epidermis, and the papillary and reticular dermis contain numerous thick-walled vessels with prominent endothelial cells. Immunohistochemically, multinucleated cells variably express CD68 and factor XIIIa, but are usually negative for CD163, CD34, and factor VIII [97,100]. Endothelial markers, including CD31, CD34, ETS-related gene (ERG), and factor VIII, highlight vascular proliferation.
EWSR1::SMAD3-rearranged fibroblastic tumor
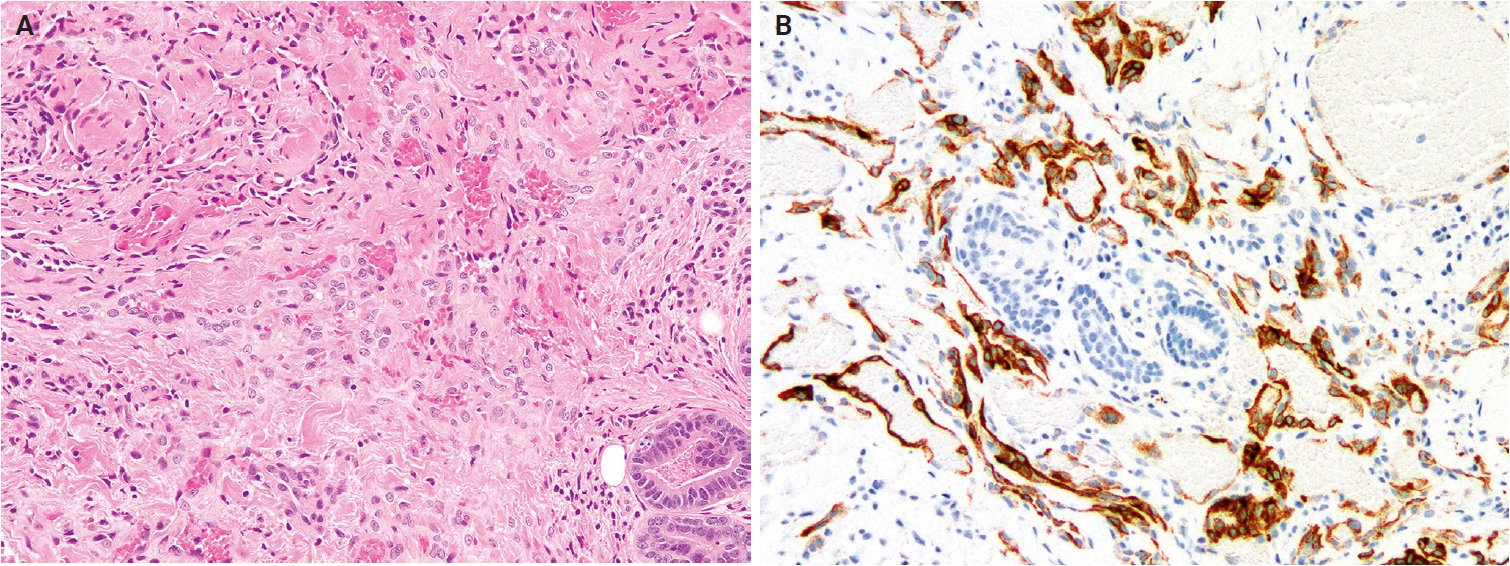
ESRFT is a rare, distinctive benign fibroblastic neoplasm [101]. Most cases arise in the acral region, particularly the hands and feet [102], although the lower extremity is the most common non-acral site. Clinically, ESRFT presents as a small, painless, superficial nodule, typically measuring 10–20 mm in diameter. It affects a wide age range (1–68 years; median age, 39 years) and shows a female predominance (male-to-female ratio, 1:4) [103]. Molecularly, ESRFT is characterized by an EWSR1::SMAD3 gene fusion, typically involving exon 7 of EWSR1 and exon 5 or 6 of SMAD3. SMAD family member 3 (SMAD3) functions as a key signal transducer in the TGF-β/SMAD pathway and is essential for fibroblast-mediated extracellular matrix synthesis, thereby supporting the fibroblastic differentiation of this tumor.
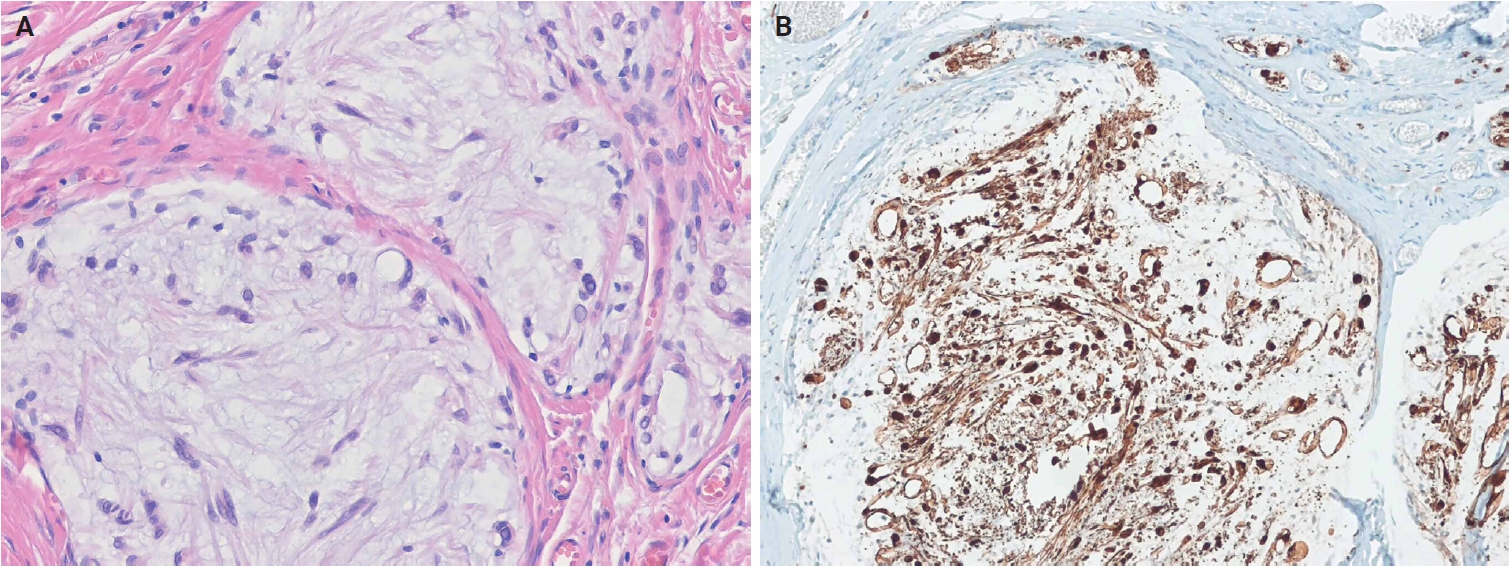
Histologically, ESRFTs are typically well-circumscribed, although focal infiltration into the subcutaneous fat may be observed. In adults, the tumor frequently exhibits a hyalinized, acellular central zone resembling collagen rosettes. This zone is surrounded by peripheral areas composed of intersecting fascicles of spindle-shaped fibroblastic cells embedded in a collagenous to variably myxoid stroma (Fig. 6A). Approximately half of the cases lack this zonation pattern. Tumor cells are cytologically bland, with no nuclear pleomorphism, hyperchromasia, prominent nucleoli, or increased mitotic activity. Myopericytomatous growth is rare, and focal stippled dystrophic calcification may be present [102,103]. Immunohistochemically, tumor cells demonstrate strong, diffuse nuclear ERG expression, whereas SMA and CD34 are negative (Fig. 6B).
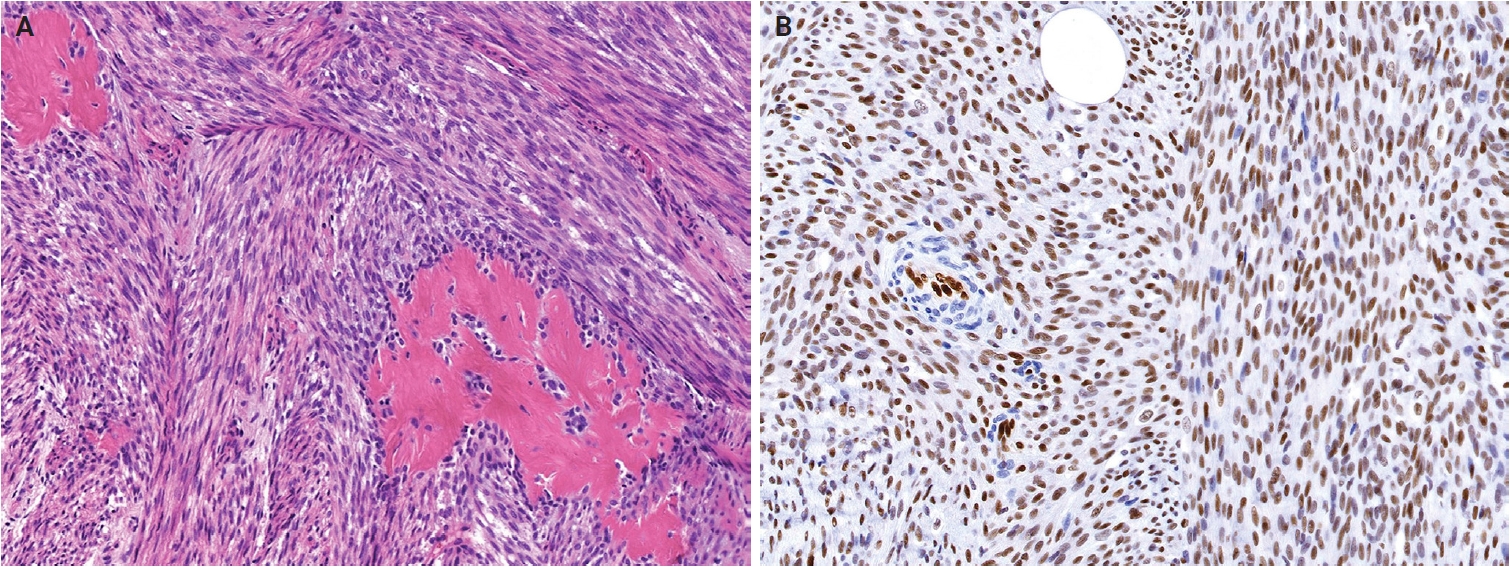
EWSR1::SMAD3-rearranged fibroblastic tumor. (A) The tumor is composed of intersecting fascicles of fibroblastic spindle cells with a hyalinized collagenous stroma. (B) The tumor cells show diffuse nuclear immunoreactivity for ERG. Reprinted from Suurmeijer et al. EWSR1::SMAD3-rearranged fibroblastic tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. [101], with permission from IARC Press.
VASCULAR TUMORS
Key updates
In the 5th WHO classification, hemangiomas and other benign vascular tumors are grouped separately. Newly described entities in the vascular tumor family include papillary hemangioma, poikilodermatous plaque-like hemangioma (PPLH), acquired elastotic hemangioma (AEH), verrucous venous malformation, and hobnail hemangioendothelioma (HHE). Tufted hemangioma, a benign cutaneous vascular tumor sometimes associated with Kasabach-Merritt syndrome, is now generally considered part of the Kaposiform hemangioendothelioma (KHE) spectrum [104,105], and KHE has been removed from this volume. The term ‘postradiation atypical vascular lesion’ replaces the previous designation ‘atypical vascular lesion’, highlighting its association with prior radiation exposure and distinguishing it from radiation-induced angiosarcoma [106]. Additionally, papillary intralymphatic angioendothelioma (PILA) and retiform hemangioendothelioma (RHE) are now unified under the designation of HHE [107].
In cherry hemangioma, somatic mutations in HRAS and KRAS have been identified in a small subset of cases [108]. The 5th edition also describes frequent somatic mutations in GNA14, GNA11, and GNAQ [109,110]. In glomeruloid hemangioma, elevated serum vascular endothelial growth factor levels suggest a potential role in lesion development [111]. In epithelioid hemangioma (EH), FOS or FOSB gene rearrangements are commonly present in bone and soft tissue EHs, although these are rare in cutaneous EH [112]. Epithelioid endothelial cells usually express FBJ murine osteosarcoma viral oncogene homolog B (FOSB) via IHC [113]. Arteriovenous malformations are now recognized as vascular malformations rather than neoplasms; terms such as arteriovenous hemangioma, cirsoid aneurysm, and acral arteriovenous tumor are no longer recommended [114].
Pseudomyogenic hemangioendothelioma (PHE) is characterized by recurrent FOSB rearrangements, with various fusion partners such as SERPINE1::FOSB, ACTB::FOSB, WWTR1::FOSB, CLTC::FOSB, and EGFL7::FOSB [115-120], resulting in overexpression of FOSB and constitutive transcriptional activity [120]. Fusion partner type may influence clinical features: tumors with ACTB::FOSB fusions are often solitary, whereas those with EGFL7::FOSB tend to be more aggressive [120]. Histologically, PHE is characterized by fascicles or sheets of spindled to epithelioid cells with vesicular nuclei and eosinophilic cytoplasm and showing expression of ERG and cytokeratin (AE1/AE3) (Fig. 7A, B). In epithelioid hemangioendothelioma (EHE), >90% of cases harbor WWTR1::CAMTA1 fusion resulting from t(1;3)(p36;q25) translocation [121,122], whereas a minor subset exhibits a YAP1::TFE3 fusion [123,124]. Nuclear expression of calmodulin-binding transcription activator 1 (CAMTA1) serves as a surrogate marker for WWTR1::CAMTA1 fusion, and nuclear transcription factor E3 (TFE3) positivity indicates YAP1::TFE3 fusion. Additionally, the loss of expression of the C-terminus of yes-associated protein 1 (YAP1) is specific to tumors with YAP1::TFE3 fusions [125]. In composite hemangioendothelioma (CHE), approximately 27% of tumors harbor YAP1::MAML2 fusions [126,127]. Neuroendocrine CHE is recognized as a distinct subtype characterized by a mixture of retiform hemangioendothelioma-like and EHE-like areas, together with a striking nested component, and by expression of neuroendocrine markers, most commonly synaptophysin (Fig. 8A, B) [126,128]. Subsets of neuroendocrine CHEs harbor PTBP1::MAML2 and EPC1::PCH2 fusions [128,129], and CHEs with YAP1::MAML2 fusions show loss of YAP1 C-terminus expression [125].
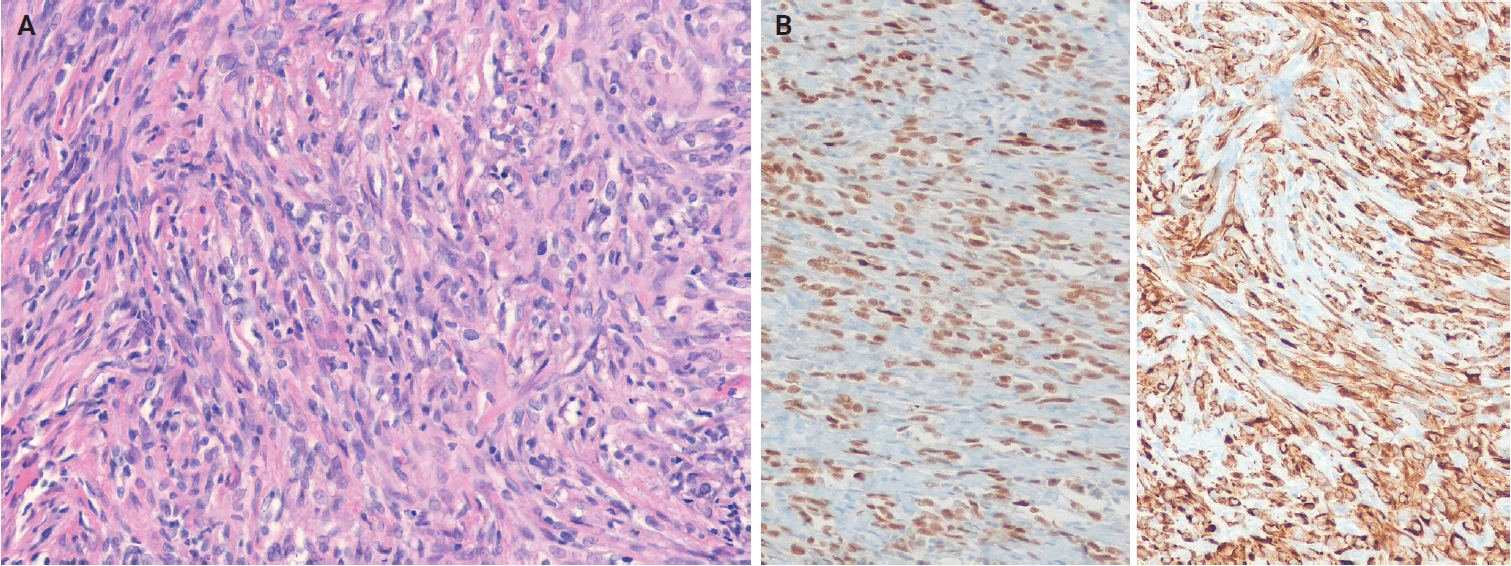
Pseudomyogenic hemangioendothelioma. (A) The tumor is composed of fascicles of spindled to epithelioid tumor cells. (B) The tumor cells show diffuse immunoreactivity for ERG (left) and cytokeratin (AE1/AE3) (right).
Neuroendocrine composite hemangioendothelioma. (A) The tumor is composed of epithelioid endothelial cells adjacent to slit-like vascular channels lined by hobnailed cells. (B) The tumor cells are positive for synaptophysin. Reprinted from Hornick et al. Composite haemangioendothelioma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. [126], with permission from IARC Press.
Kaposi sarcoma (KS) is classified into four clinical subtypes: classic, endemic African, acquired immunodeficiency syndrome–associated, and iatrogenic [130]. Genomic profiling reveals recurrent 11q13 gains with amplification of FGF4 and FGF3 (INT2) [131]. Early-stage KS shows clonal loss of the Y chromosome, whereas late (nodular) lesions accumulate copy-number alterations on chromosomes 16, 17, 21, X, and Y [132]. Cutaneous angiosarcoma demonstrates expanded genetic heterogeneity (Fig. 9A, B) [133]. Tumors arising in the head, neck, face, and scalp frequently exhibit ultraviolet (UV) radiation-associated mutation signature and high tumor mutation burden, which may have implications for immuno-oncology-based therapy [134]. MYC amplifications are nearly universal in secondary angiosarcoma (post-radiation or lymphedema-associated) but occur in <10% of primary tumors [135-137]. The most common recurrent mutations involve KDR, PTPRB, and PLCG1 (approximately 40%) [138,139], whereas mutations in RAS, PIK3CA, TP53, FLT4, and TIE1 are less frequent [140,141]. Additionally, a CIC-altered subset occurs in younger patients and is associated with poorer disease-free survival [142].
Cutaneous angiosarcoma. (A) The tumor shows numerous infiltrative vascular channels dissecting between dermal collagen bundles. (B) These anastomosing vascular channels are lined by atypical endothelial cells, with focal endothelial multilayering.
Papillary hemangioma
Papillary hemangioma is a distinctive benign vascular proliferation characterized by dilated vascular channels containing prominent intraluminal papillary projections [143]. It occurs almost exclusively in the head and neck region and predominantly affects adult men [144,145]. Clinically, it presents as a solitary, long-standing, bluish-red papule, measuring up to 11 mm in diameter.
Histologically, papillary hemangioma demonstrates widely dilated, thin-walled vascular channels in the dermis, containing numerous intraluminal papillary projections. These papillary structures comprise capillary-sized vessels lined by plump endothelial cells and surrounded by pericytes. Endothelial cells often contain intracytoplasmic eosinophilic globules (so-called ‘thanatosomes’), corresponding to dilated lysosomes containing fat vacuoles and cellular debris. Immunohistochemically, endothelial cells are positive for CD31, CD34, and ERG, but negative for lymphatic markers such as podoplanin (D2-40).
Poikilodermatous plaque-like hemangioma
PPLH is an acquired benign vascular proliferation characterized by a band-like pattern of small blood vessels in the superficial dermis [146]. Almost all lesions arise on the lower extremities [147,148], predominantly affecting older men. Clinically, PPLH presents as slowly growing, asymptomatic, erythematous to violaceous, scaly plaques, measuring 20−70 mm in diameter, often exhibiting poikilodermatous changes. Lesions are usually solitary but may be multiple. The course is indolent, and lesions tend to persist despite treatment.
Histologically, PPLH demonstrates a band-like proliferation of small-caliber blood vessels lined by bland endothelial cells within the papillary and superficial reticular dermis. The affected area shows loss or marked reduction of elastic fibers, and a grenz zone is absent. Variable degrees of hyperkeratosis, acanthosis, and mild perivascular lymphocytic infiltration are also commonly observed.
Acquired elastotic hemangioma
AEH is a rare, benign vascular proliferation characterized by a superficial, band-like arrangement of small blood vessels in sun-exposed skin [149]. It is typically associated with solar elastosis. AEH predominantly occurs on the forearms or lateral neck and presents as an asymptomatic papule or plaque, primarily affecting middle-aged to elderly adults, with a slight female predominance [150,151]. Multiple lesions are rare, and chronic sun damage is considered the primary etiologic factor.
Histologically, AEH exhibits a band-like proliferation of capillary-sized vessels, each surrounded by a layer of pericytes and oriented parallel to the epidermis within the upper reticular dermis. These vessels are set against a background of prominent solar elastosis.
Hobnail hemangioendotheliomas
PILA and RHE are uncommon, rarely metastasizing vascular neoplasms—both feature hobnail endothelial proliferations with a lymphatic endothelial phenotype [107]. PILA typically arises in the proximal extremities, particularly the buttock and thigh [152-154], whereas RHE predominantly occurs in the distal extremities, especially the lower leg [155,156]. Fewer than 50 cases of PILA and RHE have been reported [152,155,156]. PILA predominantly affects infants, with a slight female predominance, whereas RHE occurs in children or young adults, with no sex predilection. The etiology remains uncertain; some RHE cases are associated with prior radiotherapy [155], lymphedema [155,157], or cystic lymphangioma [158]. A subset of RHE harbors YAP1 rearrangements, most often YAP1::MAML2 fusions [127].
Histologically, PILA and RHE are morphologically similar, composed of darkly staining hobnail or columnar endothelial cells. PILA exhibits poorly defined proliferation of slit-like or dilated lymphatic channels, occasionally associated with cavernous lymphatic malformation. In contrast, RHE exhibits infiltrative growth of elongated, branching vascular channels resembling the rete testis. Shared features include hyaline fibrosis, intraluminal papillary tufts lined by hobnail endothelial cells—more conspicuous in PILA—and a lymphocytic infiltrate. Immunohistochemically, hobnail endothelial cells of PILA and RHE express pan-endothelial markers (CD31, ERG, Friend leukemia virus integration 1 [FLI1], and CD34) and lymphatic markers (podoplanin [D2-40], vascular endothelial growth factor receptor 3 [VEGFR3], and prospero homeobox 1 [PROX1]) [158,159]. Tumors harboring YAP1 rearrangements show loss of YAP1 C-terminal expression [151]. Focal synaptophysin expression and shared YAP1::MAML2 fusions support a pathogenetic link between HHE and CHE.
PERICYTIC AND PERIVASCULAR TUMORS
Key updates
No major revisions or newly recognized entities have been added to this family in the 5th edition. Myofibroma and myofibromatosis, previously categorized among fibroblastic, myofibroblastic, and fibrohistiocytic tumors, have been reclassified as pericytic and perivascular tumors. In glomus tumor, sporadic tumors often show rearrangements involving MIR143 and NOTCH genes [160-162], whereas BRAF p.V600E mutations have been reported in rare malignant cases [163,164]. Demonstration of MIR143::NOTCH1, MIR143::NOTCH2, or MIR143::NOTCH3 fusions can aid in diagnosis [161,162].
The 5th edition includes newly recognized multicentric and intravascular subtypes of myopericytoma [165]. Pericytoma-like neoplasms harboring recently identified gene fusions, such as ACTB::GLI and SRF::RELA, are likely unrelated to myopericytoma [166,167]. Angioleiomyomas exhibit cytogenetic abnormalities, including monosomy of chromosome 13 and losses of 6p, 21q, and 13q [168,169]; rare cases harbor NOTCH gene fusions [161,162]. In addition to solid, venous, and cavernous subtypes, hybrid subtypes have also been described. Similar to glomus tumors and myopericytomas, angioleiomyomas demonstrate diffuse immunoreactivity for the pericytic marker regulator of G-protein signaling 5 [170].
Myofibroma and myofibromatosis
Myofibroma and myofibromatosis are benign mesenchymal neoplasms of presumed myofibroblastic lineage, presenting as solitary, multicentric, or generalized lesions [171]. Solitary lesions most commonly involve the skin or subcutaneous tissue of the head and neck, upper extremities, or trunk. However, deep lesions involving muscles and bones are uncommon [172]. Multicentric lesions share a similar distribution, whereas generalized lesions involve visceral organs. Solitary lesions predominantly affect males, while multicentric lesions occur mainly in females. Myofibroma accounts for approximately 12% of pediatric soft tissue lesions, with 90% of cases occurring in children and 65% presenting before age 2. Molecularly, familial myofibromatosis exhibits germline PDGFRB mutations (approximately 89%) and NOTCH3 mutations (approximately 11%), whereas sporadic solitary lesions frequently harbor somatic PDGFRB mutations [173,174]. PDGFRB mutation types differ between familial and sporadic myofibromatosis cases, and SRF rearrangements have been reported in highly cellular tumors [175].
Histologically, myofibroma shows a biphasic growth pattern comprising alternating bundles of primitive, small, darkly staining, rounded cells and plump, spindle-shaped mesenchymal cells (Fig. 10A, B). Tumors are richly vascularized, with numerous thin-walled, hemangiopericytoma-like vessels and mature eosinophilic myoid cells, often accompanied by myointimal nodular proliferations (‘vascular balls’). Foci of calcification and ischemic-type necrosis may be present, and mitotic activity is typically low. Immunohistochemically, tumor cells are diffusely positive for SMA, while caldesmon is negative or focally positive, and desmin is generally negative.
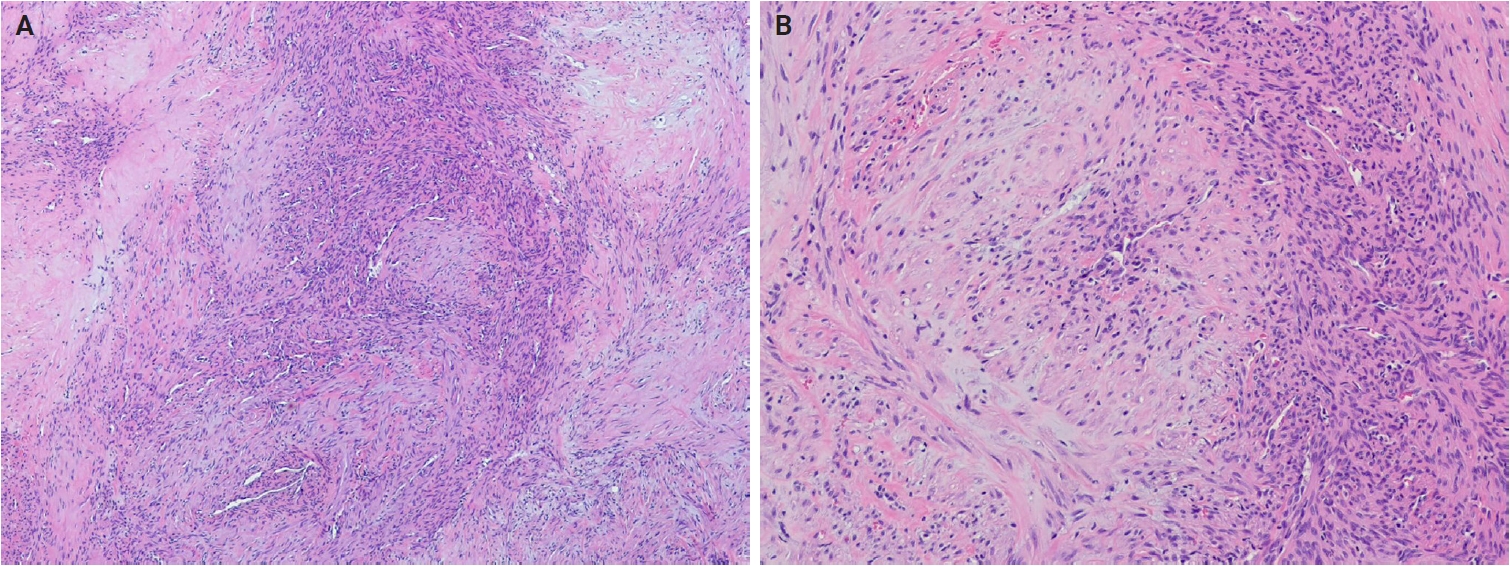
Myofibroma. (A) The tumor shows a characteristic biphasic growth pattern with ill-defined nodules of myoid cells and intervening hypercellular areas. (B) Eosinophilic spindled myoid cells and smaller ovoid cells are present around pericytoma-like vessels.
SMOOTH MUSCLE TUMORS
Key updates
No major revisions or newly recognized entities have been added to this family in the 5th WHO classification. Smooth muscle hamartoma (SMH) and Epstein-Barr virus–associated smooth muscle tumor (EBV-SMT) are newly included in the family of smooth muscle tumors. In the 5th edition, dermal-confined lesions previously termed ‘cutaneous leiomyosarcoma’ are designated as ‘atypical intradermal smooth muscle neoplasm’, whereas the term ‘cutaneous leiomyosarcoma’ is reserved for tumors with overt subcutaneous fat infiltration (Fig. 11A, B) [176]. Prognosis is excellent; no metastases have been reported in the largest series to date, comprising 84 cases of completely excised dermal-confined tumors [177]. Although data remain limited, dermal tumors with minimal subcutaneous extension have a low risk of recurrence or metastasis. Histological grading does not provide prognostic value.
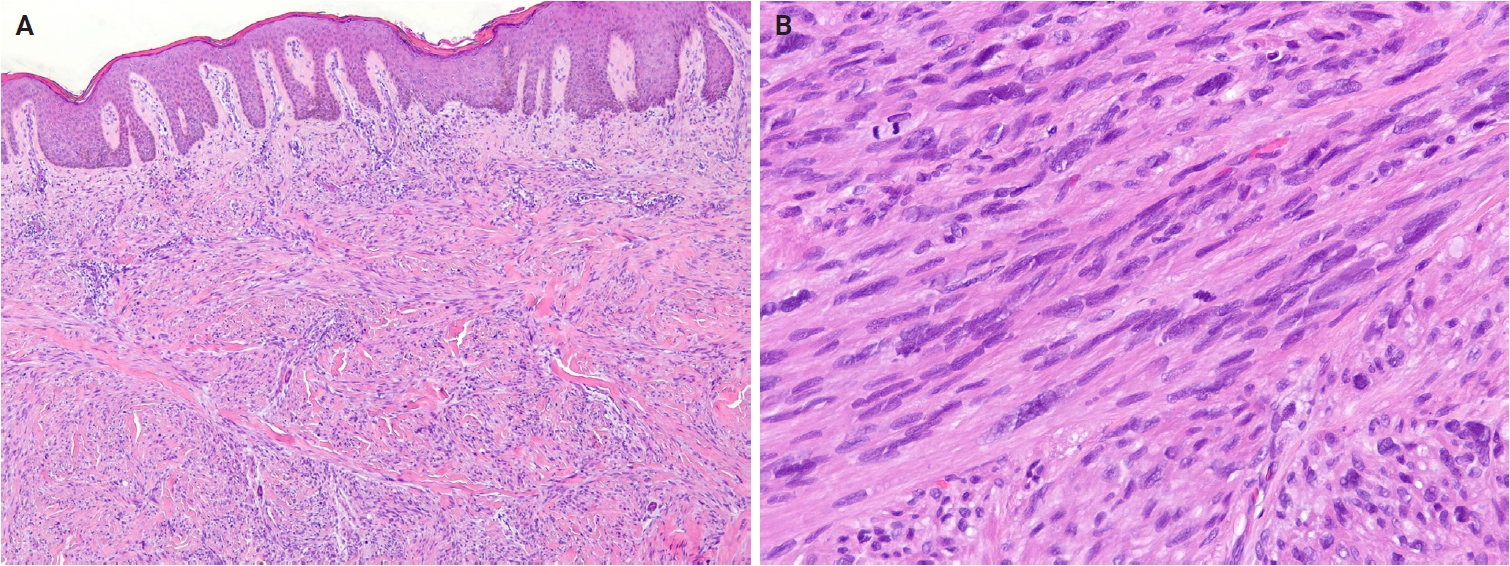
Atypical intradermal smooth muscle neoplasm. (A) The tumor is composed of infiltrative spindle cells within the dermis. (B) The tumor cells show elongated, blunt-ended nuclei and brightly eosinophilic cytoplasm. Moderate nuclear atypia and mitotic figures are present.
Smooth muscle hamartoma
SMH is a benign dermal lesion composed of haphazardly arranged smooth muscle bundles [178]. It most commonly occurs on the trunk or extremities, with less frequent involvement of the head, neck, and genital regions [179]. A positive pseudo-Darier sign is observed in up to 80% of congenital cases. Most congenital SMHs are solitary, although rare multiple, familial, or diffuse forms have been reported [180]. Diffuse congenital SMHs may be associated with Michelin tire baby syndrome [181], and combined lesions with congenital or blue nevi have also been reported [182,183]. SMH exhibits a slight male predominance, and congenital cases are uncommon, with an estimated incidence of 1 in 2,600 births [182]. Molecularly, a subset of congenital SMHs harbors postzygotic ACTB mutations, also identified in Becker nevi, suggesting a shared biological basis between these lesions [183].
Histologically, SMH exhibits haphazard bundles of mature smooth muscle within the dermis, often adjacent to pilosebaceous units [184]. Cytological atypia, pleomorphism, and mitotic activity are absent. Focal retraction artifacts may be present around smooth muscle bundles. Lesional cells express smooth muscle markers. The overlying epidermis frequently demonstrates hyperkeratosis, acanthosis, and basal hyperpigmentation, resembling features of a Becker nevus.
EBV-associated smooth muscle tumor
EBV-SMT is a rare EBV-driven smooth muscle neoplasm that develops in immunosuppressed patients [185]. The central nervous system is the most commonly affected, followed by the liver, lungs, and subcutaneous tissues of the trunk and extremities [186-189]. EBV-SMT occurs in approximately 1%–5% of immunosuppressed individuals [189], affecting a wide age range, most often adults, with a slight female predominance [190]. Tumorigenesis is driven by EBV type 2 infection in the context of a congenital immunodeficiency disorder, post-transplant immunosuppression, or human immunodeficiency virus infection [189,191]. EBV infects vascular smooth muscle progenitor cells via CD21 receptor-mediated entry or through EBV-infected lymphocytes [192,193]. Aberrant activation of the mTOR/AKT signaling pathway and MYC overexpression contribute to pathogenesis [193,194]. Multiple lesions typically represent independent clonal proliferations rather than metastases [186]. Prognosis is generally favorable and depends more on immune status than on tumor morphology [186,189].
Histologically, EBV-SMTs are well-circumscribed lesions composed of interlacing fascicles of spindle-shaped myoid cells within a collagenous stroma, irrespective of anatomical site [195,196]. A variable admixture of smaller rounded cells and intratumoral lymphocytes is typically present, and focal myxoid change may be observed. Mitotic activity is generally low, although rare tumors exhibit leiomyosarcoma-like features, including nuclear atypia, necrosis, and increased mitotic activity [190,191]. Immunohistochemically, tumor cells express smooth muscle markers, including SMA, muscle-specific actin, desmin, and caldesmon. EBV infection can be confirmed by in situ hybridization test for Epstein-Barr virus-encoded small RNA.
NEURAL TUMORS
Key updates
In the 5th edition, dermal hyperneury (DHN)/epithelial sheath neuroma (ESN) and hybrid nerve sheath tumor have been newly described within the neural tumors family. In solitary circumscribed neuroma (SCN), the plexiform subtype has been newly recognized as part of the SCN spectrum [197]. Dermal nerve sheath myxoma (DNSM) is now defined as a benign peripheral nerve sheath tumor (Fig. 12A, B) [198]. Pathological and molecular evidence supports its Schwann cell origin [198-201]. Gene expression profiling of DNSM aligns with dermal schwannoma, whereas cellular neurothekeoma exhibits molecular similarities to cellular dermatofibroma (fibrous histiocytoma) [201].
Dermal nerve sheath myxoma. (A) The tumor shows a multinodular growth pattern separated by fibrous septa, with spindle, stellate, and ring-shaped tumor cells in prominent myxoid stroma. (B) The tumor cells are positive for S100 protein.
Three subtypes of perineurioma are recognized: reticular, sclerosing, and malignant [202]. Perineuriomas share pathogenetic features with schwannomas and meningiomas, including 22q12 deletion, monosomies, and NF2 mutations [203-205]. Soft tissue perineuriomas commonly exhibit a 17q11 deletion involving the NF1 locus. The sclerosing subtype demonstrates a distinctive 10q24 rearrangement [206]. Malignant perineuriomas are associated with loss of chromosome 13q and small deletions on chromosomes 3, 6, and 9 [205,207].
Neurofibroma formally recognizes melanotic neurofibroma as a distinct subtype [208]. Multiple tumors, particularly plexiform and diffuse subtypes, are strongly associated with neurofibromatosis type 1 (NF1) and type 2 (NF2). Genotype–phenotype correlations have been established in NF1 [209]. Loss of CDKN2A at chromosome 9p21.3 may contribute to the development of atypical neurofibromatous tumor of uncertain biological potential [210]. Schwannoma is classified into six subtypes: plexiform, epithelioid, ancient schwannoma, cellular, microcystic/reticular, and neuroblastoma-like [211].
Granular cell tumor (GCT) is a benign neuroectodermal tumor derived from Schwann cells [212]. Loss-of-function mutations in ATP6AP1 and ATP6AP2, which are mutually exclusive, occur in approximately 70% of cases [213] and are considered pathognomonic of GCT. Malignant GCT is characterized by overt malignant features, including increased cellularity, prominent spindling, a high nuclear-to-cytoplasmic ratio, marked pleomorphism, elevated mitotic activity (including atypical figures), necrosis, and a high Ki-67 proliferation index [214-216].
Cutaneous malignant peripheral nerve sheath tumor (MPNST) is rare [217]. Unlike deep MPNSTs, cutaneous MPNSTs are less frequently associated with NF1. Conventional deep-seated MPNSTs harbor inactivating mutations in NF1, CDKN2A/CDKN2B, and polycomb repressive complex 2 (PRC2) core components (EED or SUZ12), resulting in complete loss of PRC2 function [218-220]. Approximately 80% of high-grade MPNSTs show PRC2 inactivation, with loss of histone H3 lysine 27 trimethylation (H3K27me3) expression. The loss of H3K27me3 is a valuable diagnostic marker for these tumors [221,222]. In contrast, epithelioid MPNSTs represent a molecularly distinct subtype, characterized by SMARCB1 inactivation in approximately 75% of cases [223]. Mutations in NF1, CDKN2A/CDKN2B, or PRC2 are uncommon in this subtype [218,224].
Dermal hyperneury/epithelial sheath neuroma
DHN is a form of small nerve hypertrophy characterized by large, prominent dermal nerve fibers [225]. ESN shows similar histological features, with mature squamous epithelium surrounding the perineurium. DHN/ESN is classified into syndromic and sporadic subtypes. Sporadic DHN shows a slight predilection for the trunk [226], whereas ESN most commonly arises on the back [227]. Sporadic lesions typically present in the seventh decade of life, whereas syndromic mucocutaneous neural proliferations may occur in patients as young as 5 years [228]. Mucocutaneous neural proliferations have been described in multiple endocrine neoplasia type 2B (MEN2B) [229], MEN2A [230], NF2 [231], and Cowden disease [232]. ESN occurs exclusively as a sporadic lesion. Pathogenesis may involve PTEN- and rearranged during transfection (RET)–mediated signaling pathways, particularly in syndromic cases [233].
Histologically, both DHN and ESN demonstrate aggregated neural tissue with enlarged nerve fibers located within the dermis or at the dermal-subcutaneous junction. Morphology is consistent between syndromic and sporadic cases. Nerve bundles are typically 1.5–4 times larger than normal and may exhibit mild neuroma-like disorganization. Occasionally, perineurial lymphoplasmacytic or histiocytic inflammation and intraneural mucin deposition are observed. ESN exhibits mature squamous epithelium forming an epithelial sheath around enlarged nerve fibers. Subcutaneous extension is rare [234].
Hybrid nerve sheath tumors
Hybrid nerve sheath tumors are rare benign peripheral nerve sheath tumors that exhibit combined features of two or more conventional types, most commonly schwannoma, neurofibroma, or perineurioma [235]. They occur across a wide anatomical range, most frequently in the dermis or subcutaneous tissue [236,237]. The most common subtype is hybrid schwannoma/perineurioma, which predominantly arises in the fingers [238]. Hybrid schwannoma/perineurioma usually occurs sporadically, whereas hybrid neurofibroma/schwannoma exhibits a striking association with NF1 and NF2 [239]. A high prevalence of hybrid schwannoma/neurofibroma morphology was found in tumors from patients with schwannomatosis (71%) [239,240]. Hybrid neurofibroma/perineurioma has also been reported in association with NF1 [241,242]. Most hybrid schwannoma/perineuriomas harbor VGLL3 gene rearrangements, supporting their classification as a distinct pathological entity rather than a simple histological overlap [243].
Histologically, a hybrid schwannoma/perineurioma exhibits an intimate admixture of alternating Schwann cells and perineurial cells. Schwann cells have wavy, tapering nuclei and eosinophilic cytoplasm, whereas perineurial cells have slender nuclei and delicate, elongated cytoplasmic processes. The Schwann cell component is usually predominant and may exhibit degenerative nuclear atypia (‘ancient change’), while mitoses are rare. Hybrid neurofibroma/schwannoma is characterized by schwannomatous nodules or bundles of Schwann cells within an otherwise typical neurofibroma. The neurofibromatous areas contain a mixture of fibroblasts, Schwann cells, and perineurial cells [242,244]. Hybrid neurofibroma/perineurioma shows plexiform neurofibroma with foci of perineurial differentiation, which are often subtle and require IHC for confirmation. Immunohistochemically, Schwann cells are positive for S100 protein and SRY-box transcription factor 10 (SOX10), whereas perineurial cells express epithelial membrane antigen (EMA), claudin-1, and GLUT1, and fibroblasts are positive for CD34.
TUMORS OF UNCERTAIN DIFFERENTIATION
Key updates
The 5th edition introduces several newly described entities in the family of tumors of uncertain differentiation, including perivascular epithelioid cell tumor (PEComa), angiomatoid fibrous histiocytoma (AFH), neurotrophic tyrosine receptor kinase (NTRK)–rearranged spindle cell neoplasm (NRSCN), superficial CD34-positive fibroblastic tumor (SCFT), and CRTC1::TRIM11 cutaneous tumor (CTCT). The SCFT, previously classified as a fibroblastic and myofibroblastic tumor in the 5th edition of the WHO classification of soft tissue tumors, has also been reclassified as a tumor of uncertain differentiation.
Cellular neurothekeoma is a benign cutaneous neoplasm of uncertain histogenesis, distinct from nerve sheath myxoma (previously called myxoid neurothekeoma) [245]. The most consistently expressed markers include CD63 (NKI/C3) [246,247], neuron-specific enolase [248], protein gene product 9.5 (PGP9.5) [249], and S100 calcium-binding protein A6 (S100A6) [250]. The 5th edition expands the immunoprofile to include keratinocyte basal antigen 62 (KBA.62) and preferentially expressed antigen in melanoma (PRAME) [251,252]. Non-neural granular cell tumor is a rare, low-grade neoplasm of mesenchymal cells of unknown lineage, characterized by prominent cytoplasmic granularity [253]. ALK rearrangements have been reported in 60% of cases in a small series [254]. Atypical fibroxanthoma is defined as a dermally confined, low-grade neoplasm of uncertain histogenesis [255]. Genomic alterations include deletions at 9p and 13q, TERT mutations, and a variety of abnormalities similar to pleomorphic dermal sarcoma (PDS) [256-258]. Morphologic patterns include spindle cell, clear cell, granular cell, keloidal, myxoid, sclerotic, and pigmented forms [259-261].
PDS is a rare tumor affecting older adults with chronically UV radiation-exposed skin [262]. It is characterized by a high UV-related mutation burden, including mutations in TP53, NOTCH family genes, the TERT promoter, and alterations in CDKN2A and CDKN2B [256,258]. This mutation profile parallels that of cutaneous squamous cell carcinoma. Immunohistochemically, PDS commonly expresses CD10, CD99, and platelet-derived growth factor receptor beta (PDGFRB).
Epithelioid sarcoma (ES) is a malignant soft tissue tumor that exhibits hybrid mesenchymal and epithelial morphology and immunophenotypes [263]. Loss of nuclear SMARCB1 (INI1) expression is characteristic of ES; however, a subset retains SMARCB1 (INI1) and instead shows aberrant expression of other SWI/SNF complex components, including SMARCA4 (BRG1), SMARCC1 (BAF155), and SMARCC2 (BAF170) [264]. Genomic analyses have revealed complex copy-number aberrations and a relatively high mutational burden [265].
Dermal clear cell sarcoma (CCS) is a malignant neoplasm of uncertain histogenesis with melanocytic differentiation, defined by EWSR1::ATF1 or EWSR1::CREB1 gene fusion [266]. Approximately 70%–90% of CCSs harbor an EWSR1::ATF1 fusion resulting from t(12;22)(q13;q12). Identification of EWSR1 rearrangement and/or confirmation of these fusions aids diagnosis, particularly in distinguishing CCS from malignant melanoma. In contrast to malignant melanomas, PRAME expression in CCS is typically absent or limited to focal areas [267,268].
Ewing sarcoma is a small round cell sarcoma defined by gene fusions involving a FUS-EWSR1-TAF15 (FET) family gene, most commonly EWSR1, and an erythroblast transformation-specific (ETS) family transcription factor [269]. All cases harbor FET::ETS fusions, which constitute the defining molecular hallmark. The tumorigenesis of cutaneous Ewing sarcoma is driven by oncogenic transcription factors encoded by chimeric FET::ETS fusion genes, most frequently EWSR1::FLI1 (>95%), and, less commonly, EWSR1::ERG [270-272].
Epithelioid fibrous histiocytoma
EFH is a distinctive, benign cutaneous neoplasm composed of epithelioid cells [273]. It most commonly arises in the lower extremities and, less frequently, in the upper extremities, trunk, or head and neck [274,275]. EFH typically occurs in young to middle-aged adults (median age, 40 years), with a female predominance. Molecularly, approximately 90% of EFHs harbor ALK rearrangements, while a small subset shows mutually exclusive PRKC gene fusions [276,277]. SQSTM1 and VCL are the most common ALK fusion partners, whereas DCTN1, ETV6, PPFIBP1, SPECC1L, TPM3, PRKAR2A, MLPH, CLTC, and EML4 represent rare fusion partners [278-281].
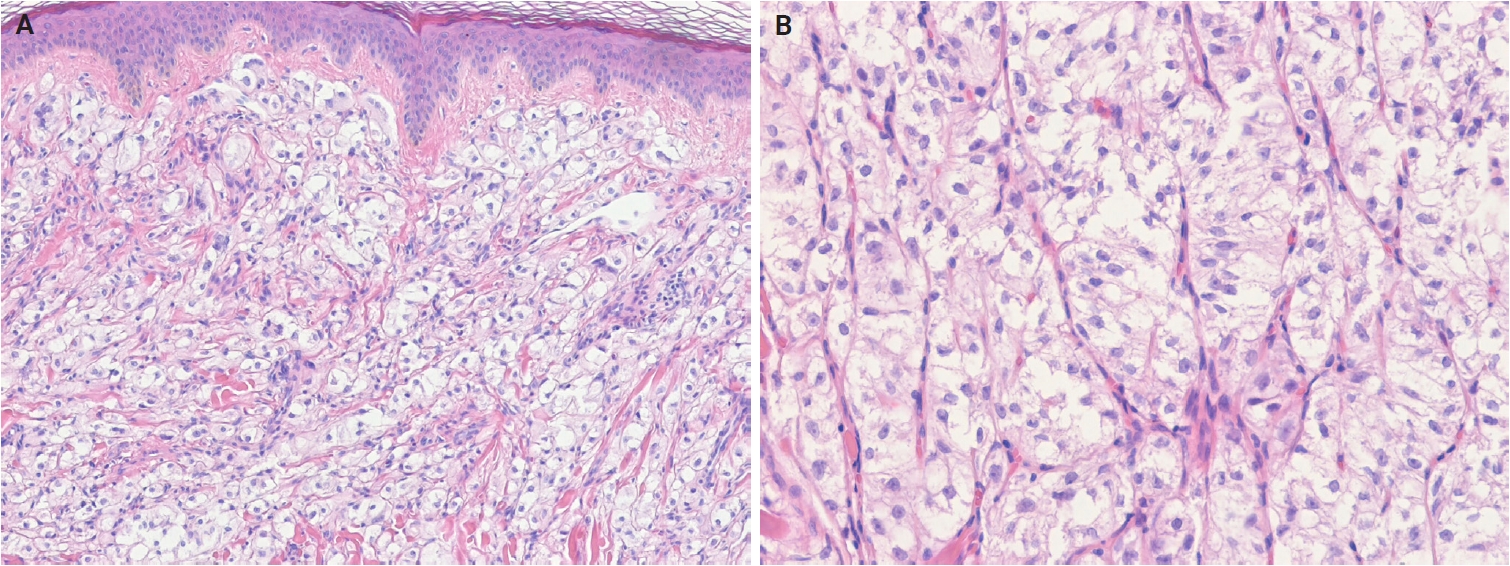
Histologically, EFH is typically well-circumscribed, and exophytic lesions often exhibit an adnexal collarette (Fig. 13A). The tumor consists of uniform epithelioid cells with vesicular nuclei, small nucleoli, and pale eosinophilic or amphophilic cytoplasm (Fig. 13B). Binucleated forms are frequently observed. Thin-walled vessels are common, often with perivascular accentuation of tumor cells. Morphological variants include chondroblastoma-like pericellular calcification [279,282] and spindle cell-predominant forms [283,284]. Immunohistochemically, ALK is overexpressed in approximately 90% of cases [285], with D5F3 or 5A4 clones demonstrating higher sensitivity than the ALK1 clone [276,286]. EMA is positive in approximately 65% of cases [286], and CD30 expression may also be present.
Epithelioid fibrous histiocytoma. (A) Scanning magnification shows a well-circumscribed, dome-shaped dermal tumor with an adnexal collarette. (B) The tumor is composed of epithelioid cells with round to oval vesicular nuclei, small nucleoli, and abundant eosinophilic or amphophilic cytoplasm. Reprinted from Requena and Hornick. Epithelioid fibrous histiocytoma. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. [273], with permission from IARC Press.
PEComa
PEComas are mesenchymal neoplasms composed of distinctive perivascular epithelioid cells that coexpress melanocytic and smooth muscle markers [287]. Cutaneous PEComas are rare and most commonly involve the lower extremities, with less frequent occurrence on the trunk, head, or neck [288-290]. They affect individuals across a wide age range (15–81 years), with a peak incidence in middle age, and show a female predominance [288,289]. Most cases are sporadic; however, fibroma-like cutaneous PEComa associated with tuberous sclerosis has been reported [291]. Molecularly, deletion of 16p involving the TSC2 gene is frequent, and loss of heterozygosity at the TSC2 locus is seen in both sporadic and syndromic tumors [292]. TP53 mutations occur in approximately 63% of TSC2-mutated PEComas [293].
Histologically, PEComa is composed of epithelioid cells with granular, pale eosinophilic, or clear cytoplasm and round nuclei with small nucleoli (Fig. 14A, B). Most PEComas exhibit a nested or trabecular architecture with a distinctive perivascular growth pattern. Mixed epithelioid-spindle cell morphology and scattered multinucleated cells may occur; however, mitoses are generally rare. Most reported cases of cutaneous PEComa lack malignant features, such as marked pleomorphism, high mitotic activity, and necrosis [292-296]. Malignant primary cutaneous PEComas are exceedingly rare [290,297]. Currently, cutaneous angiomyolipomas are classified as hamartomatous or lipomatous neoplasms with smooth muscle elements, rather than true PEComas. Immunohistochemically, PEComas coexpress melanocytic markers (e.g., human melanoma black 45 [HMB-45], melan-A (MART1), microphthalmia-associated transcription factor [MITF]) and muscle markers (e.g., SMA, desmin, caldesmon) [298,299].
Perivascular epithelioid cell tumor. (A) The tumor shows dermal epithelioid cells arranged in a trabecular and nested growth pattern. (B) The tumor cells display round to oval nuclei with fine chromatin and abundant clear to pale eosinophilic cytoplasm, with a delicate capillary network surrounding the tumor nests.
Angiomatoid fibrous histiocytoma
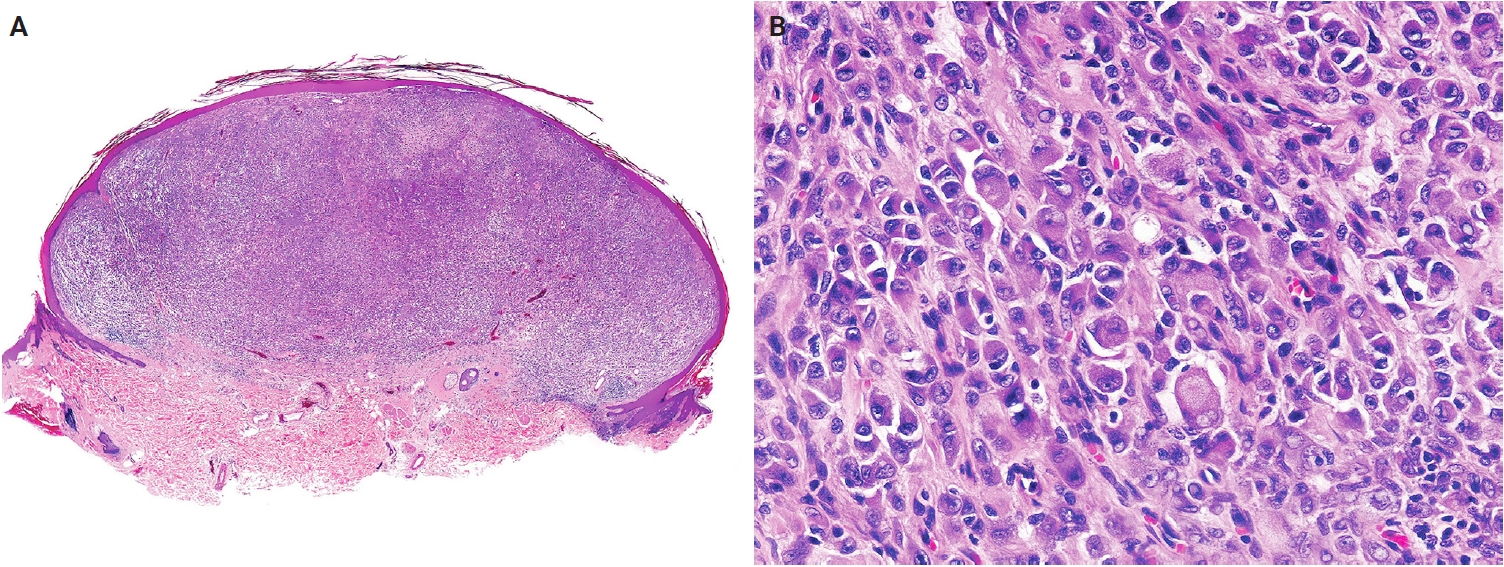
AFH is a rare soft tissue neoplasm of intermediate malignant potential and uncertain differentiation [300]. It accounts for approximately 0.3% of all soft tissue tumors. It typically presents as a dermal or subcutaneous lesion, most commonly in the extremities, followed by the trunk, head, and neck regions. Approximately two-thirds of cases arise in sites where lymph nodes are normally present [301]. AFH affects a wide age range, but most commonly presents in the first two decades of life [302-304] and shows no sex predilection. Molecularly, the characteristic alteration is t(2;22)(q33;q12), resulting in an EWSR1::CREB1 fusion, followed by t(12;22)(q12;q12), resulting in an EWSR1::ATF1 fusion [305,306]. Rare cases harbor FUS::ATF1 fusion [307,308].
Histologically, AFH consists of fascicles or sheets of oval to spindle-shaped cells with bland vesicular nuclei and prominent pseudoangiomatous blood-filled spaces. The tumor is characterized by a fibrous pseudocapsule with hemosiderin deposition and a pericapsular rim of lymphoplasmacytic infiltrates containing germinal centers, which may mimic a metastatic lymph node. Mitotic figures are usually infrequent, although atypical mitoses and pleomorphic cells may be present [309,310]. Two histological subtypes have been described: a small round cell subtype resembling undifferentiated round cell sarcoma [301,311] and a myxoid subtype [312,313]. Immunohistochemically, most AFHs express SRY-box transcription factor 9 (SOX9) [314], and approximately 50% are positive for desmin. EMA, CD99, and CD68 demonstrate variable immunoreactivity [301]. The Ki-67 proliferation index is typically low [313].
NTRK-rearranged spindle cell neoplasm
NRSCN is a recently recognized molecularly defined spindle cell tumor characterized by activating NTRK or other kinase fusions [315]. These tumors typically arise in the deep dermis and subcutis of the extremities, trunk, and head and neck area [316-318]. Although most cases occur in children and young adults, the age range is wide (0–77 years; median age, 19 years), with no sex predilection [318,319]. Oncogenesis is driven by mitogen-activated protein kinase pathway activation via kinase fusions, most commonly involving LMNA::NTRK1, TPR::NTRK1, or TPM3::NTRK1 fusions [315,316]. Alternative rearrangements involving NTRK2, NTRK3, RAF1, BRAF, RET, MET, ROS1, or ALK have also been reported [318]. NRSCNs are locally infiltrative and prone to local recurrence after incomplete excision. However, metastasis is rare, and the prognostic relevance of histologic grade remains uncertain [318,319].
Histologically, NRSCNs exhibit a broad morphological spectrum. Superficial lesions often show a lipofibromatosis-like pattern, characterized by fibrous septa and spindle cells with ovoid to tapering nuclei and indistinct cytoplasm extending through the adipose tissue (Fig. 15A). Some tumors may be more cellular, with spindled to ovoid cells arranged in fascicles or dispersed loosely in a myxoid stroma. Tumors commonly exhibit hybrid patterns, with highly cellular areas devoid of entrapped adipose tissue admixed with adipose-rich regions. Additionally, lymphocytic infiltrates, staghorn-like vessels, perivascular hyalinization, and stromal collagen bands may also be present. High-grade features such as marked atypia, high mitotic activity, or necrosis are infrequently observed [318,319]. Immunohistochemically, NRSCNs typically coexpress CD34 and S100 protein, with occasional SMA positivity. Tumors with activating NTRK fusions show diffuse pan–tropomyosin receptor kinase (pan-TRK) expression (Fig. 15B) [319,320], but the immunoprofile may be non-specific [316,318]. Therefore, molecular testing is necessary for a definitive diagnosis and for selecting targeted therapy.
NTRK-rearranged spindle cell neoplasm. (A) A tumor with a TPR::NTRK1 fusion shows spindle cells infiltrating through subcutaneous fat in a lipofibromatosis-like pattern. (B) A tumor with a TPM3::NTRK1 fusion shows diffuse immunoreactivity for pan-TRK. Reprinted from Suurmeijer and Davis. NTRK-rearranged spindle cell neoplasm. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. [315], with permission from IARC Press.
Superficial CD34-positive fibroblastic tumor
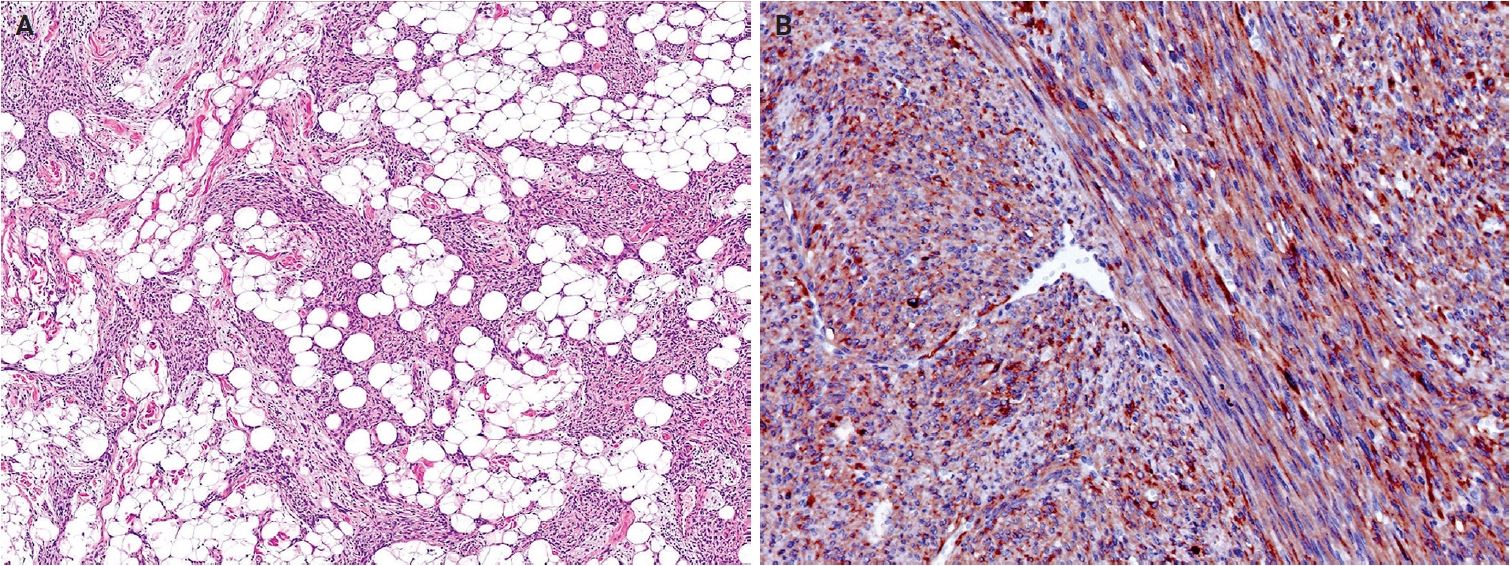
SCFT is a distinctive, low-grade neoplasm of the skin and subcutaneous tissue, characterized by a consistent morphology and diffuse CD34 expression [321]. It predominantly arises in the lower extremities, particularly the thigh, and less frequently, in the arm, buttock, shoulder, and vulva [322-325]. SCFT typically affects middle-aged adults (median age, 38 years) and shows a slight male predominance [324-327]. Morphological, immunophenotypic, and molecular findings are identical to those of ‘PRDM10-rearranged soft tissue tumor’, supporting their classification as a single entity [325,328]. However, the term ‘SCFT’ is retained because not all tumors demonstrate detectable PRDM10 fusions. PRDM10 rearrangements have been identified in approximately 60% of cases previously classified as SCFT or PRDM10-rearranged soft tissue tumors [325,328]. The prognosis is excellent [323,324].
Histologically, SCFTs are relatively well-circumscribed but partially infiltrative, composed of spindled to pleomorphic cells arranged in fascicles or sheets (Fig. 16A). Tumor cells have abundant eosinophilic cytoplasm, often granular or glassy, and lipidized cells are frequently observed. Nuclear pleomorphism ranges from moderate to marked, with bizarre, hyperchromatic nuclei, prominent nucleoli, and occasional intranuclear cytoplasmic pseudoinclusions. Mitotic activity is low, and necrosis is rare [322,323]. Mixed inflammatory infiltrates are common, and myxoid stroma or metaplastic bone may rarely be present [324]. Immunohistochemically, SCFTs exhibit diffuse CD34 expression and focal cytokeratin (AE1/AE3) positivity in approximately 70% of cases (Fig. 16B). The Ki-67 proliferation index is low (<5%) [324,325]. PRDM10 is consistently expressed [325], and CADM3 (SynCam3) expression has been reported in 95%–100% of cases [324,325].
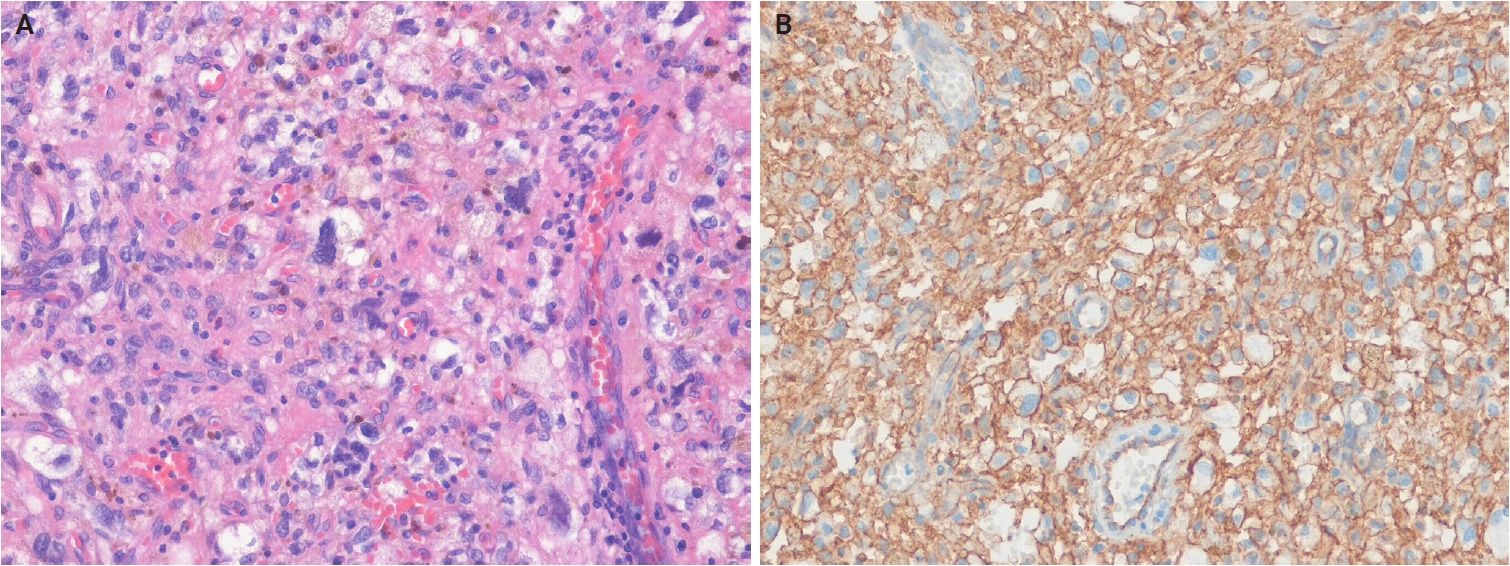
Superficial CD34-positive fibroblastic tumor. (A) The tumor is composed of spindle to pleomorphic tumor cells with an associated inflammatory infiltrate. (B) The tumor cells show diffuse CD34 expression.
CRTC1::TRIM11 cutaneous tumors
CTCT is a dermal neoplasm composed of spindle and epithelioid cells with partial melanocytic differentiation, defined by the characteristic CRTC1::TRIM11 fusion [329]. Recently recognized MITF pathway–activated tumors with melanocytic differentiation include three neoplasms defined by ACT::MITF, MITF::CREM, and CRTC1::TRIM11 fusions. In the current WHO classification, ACT::MITF- and MITF::CREM-rearranged tumors are grouped as MITF pathway–activated melanocytic tumors within the melanocytic neoplasms family [330], whereas CTCTs are classified as soft tissue tumors of uncertain differentiation. CTCTs arise at various anatomical sites, most commonly in the extremities (upper, 40%; lower, 30%), followed by the trunk (15%) and, less frequently, the head or mucosal regions (oral and nasal) [331,332]. Approximately 50 cases of CTCT have been reported, with no sex predilection and an age range of 11–87 years (median age, 44 years) [331-338]. The molecular role of CRTC1::TRIM11 fusion protein remains unclear; CRTC1 likely contributes to cell proliferation and differentiation by activating cAMP response element-binding protein 1 (CREB1), which subsequently upregulates MITF expression [339-341]. Clinically, CTCTs are usually indolent; however, local recurrence or metastasis occurs in approximately 10% of cases after excision. Confirmation of the CRTC1::TRIM11 fusion is required for diagnosis.
Histologically, CTCTs are well-circumscribed, unencapsulated, dermal-based nodules that lack pigment and may resemble CCS. Tumors are composed of nests and intersecting fascicles of spindle to epithelioid cells with vesicular nuclei, prominent nucleoli, and abundant pale cytoplasm (Fig. 17A). Mitoses are present, but infrequent, and epidermal involvement is rare. Immunohistochemically, CTCTs show diffuse SOX10 positivity (Fig. 17B). Approximately 50% of cases exhibit diffuse S100 protein staining, while melan-A (MART1) and HMB-45 display focal expression. Pan-TRK expression is present in approximately 60% of cases, and TRIM11 shows nuclear positivity at the tumor periphery [335-337]. The differential diagnoses include CCS and metastatic malignant melanoma, and definitive diagnosis requires confirmation of CRTC1::TRIM11 fusion.
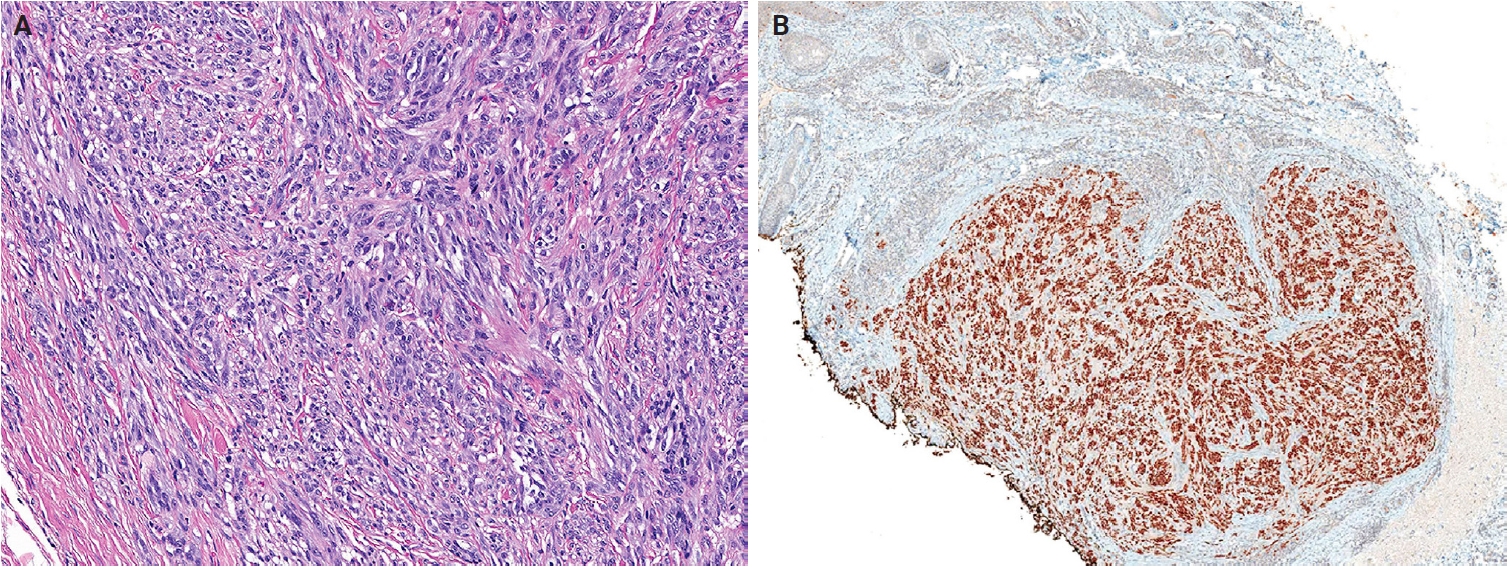
CRTC1::TRIM11 cutaneous tumor. (A) The tumor is composed of nests and long, intersecting fascicles of spindled and epithelioid cells with mild atypia in a fibrous stroma. (B) The tumor cells show diffuse nuclear positivity for SOX10. The dermal tumor is well-circumscribed and unencapsulated. Reprinted from de la Fouchardiere and Ko. CRTC1::TRIM11 cutaneous tumour. In: WHO Classification of Tumours Editorial Board, ed. WHO classification of tumours: skin tumours. 5th ed. [329], with permission from IARC Press.
CONCLUSION
This review summarizes key updates and newly recognized entities of cutaneous soft tissue tumors included in the 5th edition of the WHO classification of skin tumors. This edition incorporates recent advances in the taxonomy of cutaneous soft tissue tumors and deepens understanding of their pathogenesis by integrating histological and molecular findings. Accurate diagnosis of cutaneous soft tissue tumors requires careful correlation of clinical, microscopic, immunohistochemical, and molecular findings, together with strict adherence to well-defined diagnostic criteria. Further studies are warranted to elucidate molecular pathogenesis and refine the classification of previously unrecognized cutaneous neoplasms.
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Conflicts of Interest
J.H.C., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article.
Funding Statement
No funding to declare.