Central nervous system tumors with BCOR internal tandem duplications: a systematic review of clinical, radiological, and pathological features in 69 cases
Article information

Abstract
Central nervous system tumors with BCL6 corepressor (BCOR) internal tandem duplications (ITDs) constitute a rare, recently characterized pediatric neoplasm with distinct molecular and histopathological features. To date, 69 cases have been documented in the literature, including our institutional case. These neoplasms predominantly occur in young children, with the cerebellum representing the most frequent anatomical location. Radiologically, these tumors present as large, well-circumscribed masses frequently demonstrating necrosis, hemorrhage, and heterogeneous enhancement. Histologically, they are characterized by a monomorphic cellular population featuring ependymoma-like perivascular pseudorosettes, myxoid stroma, and elevated mitotic activity. Immunohistochemically, these tumors exhibit sparse glial fibrillary acidic protein expression while consistently demonstrating positive staining for vimentin and CD56. The defining molecular hallmark is a heterozygous ITD within exon 15 of the BCOR gene, with insertions ranging from 9 to 42 amino acids in length. BCOR immunohistochemistry reveals nuclear positivity in 97.9% of examined cases, although this finding is not pathognomonic for BCOR ITDs. This comprehensive review synthesizes data from all published cases of this novel tumor entity, providing a detailed analysis of clinical presentation, neuroimaging findings, histopathological features with differential diagnostic considerations, therapeutic approaches, and prognostic outcomes.
INTRODUCTION
Central nervous system (CNS) tumors harboring internal tandem duplications (ITDs) in the BCL6 corepressor (BCOR) gene constitute a recently recognized rare pediatric neoplasm [1]. Previously misdiagnosed due to histological resemblance to various tumor types, these neoplasms have now been established as a distinct molecular entity based on their characteristic genetic alterations, histopathological features, and immunophenotypic profiles [2]. Although awareness of this tumor type has increased substantially in recent years, fundamental questions persist regarding their precise nosological classification, diagnostic criteria, and clinical behavior. This comprehensive review synthesizes and critically evaluates the current understanding of the clinicopathologic spectrum of CNS tumors with BCOR ITD, encompassing their molecular foundations, radiologic characteristics, histologic diversity, immunohistochemical profiles, and prognostic significance.
EPIDEMIOLOGY AND CLINICAL FEATURES
This review encompasses 68 cases documented in the literature in addition to our institutional case, totaling 69 cases [1-18]. The detailed information for the 69 cases analyzed in this review article is summarized in Supplementary Table S1. The clinical information from our institutional case was approved by the Institutional Review Board of Chonnam National University Hwasun Hospital (CNUHH-2025-102). These neoplasms predominantly affect young children, with a mean age of 4.7 years and median age of 3.2 years (range, 0 to 22 years). Among the 62 cases with specified sex (excluding 7 cases with unreported sex), there is no significant sex predilection, with a male-to-female ratio of 1.13:1 (33:29). Currently, no specific environmental or hereditary risk factors have been identified for CNS tumors with BCOR ITDs.
The clinical presentation is typically characterized by symptoms secondary to elevated intracranial pressure, including headache and vomiting. Additional clinical manifestations documented in the literature include visual disturbances, ataxia, seizures, torticollis, dysphagia, hemiparesis, hand contracture, weight loss, and anorexia.
Tumor location
These tumors have been identified in various CNS locations. Among the total of 69 reported cases, including our institutional case, tumor location was specified in 65 cases. Of these, 36 cases occurred in infratentorial locations, with the cerebellum being the most frequent site (28 cases, 43.1%). The remaining eight infratentorial cases were distributed in other posterior fossa regions, including one case each in the fourth ventricle, brainstem, and cerebellopontine angle. Among the 29 cases arising in supratentorial locations, 27 cases involved various neocortical regions, while one case each occurred in the ventricular septum and basal ganglia. Among the 41 cases with available data on tumor multiplicity, all presented as solitary lesions.
RADIOLOGIC FEATURES
Radiologically, these tumors typically present as large, well-circumscribed masses with associated mass effect. Tumor diameters range from 3.8 to 10.2 cm. Central necrosis and hemorrhagic components are common, while contrast enhancement patterns are variable but generally lack the ring-enhancing pattern characteristic of glioblastoma. On diffusion-weighted imaging, restricted diffusion suggests high cellularity. Combined analysis of a cohort (10 cases) [2] with 59 additional cases from multiple case reports and the present case confirms the predominance of well-demarcated solid and cystic masses, often associated with hemorrhage or calcification (Fig. 1). Post-gadolinium magnetic resonance imaging reveals heterogeneous enhancement in most tumors, frequently exceeding 5 cm in maximal diameter.
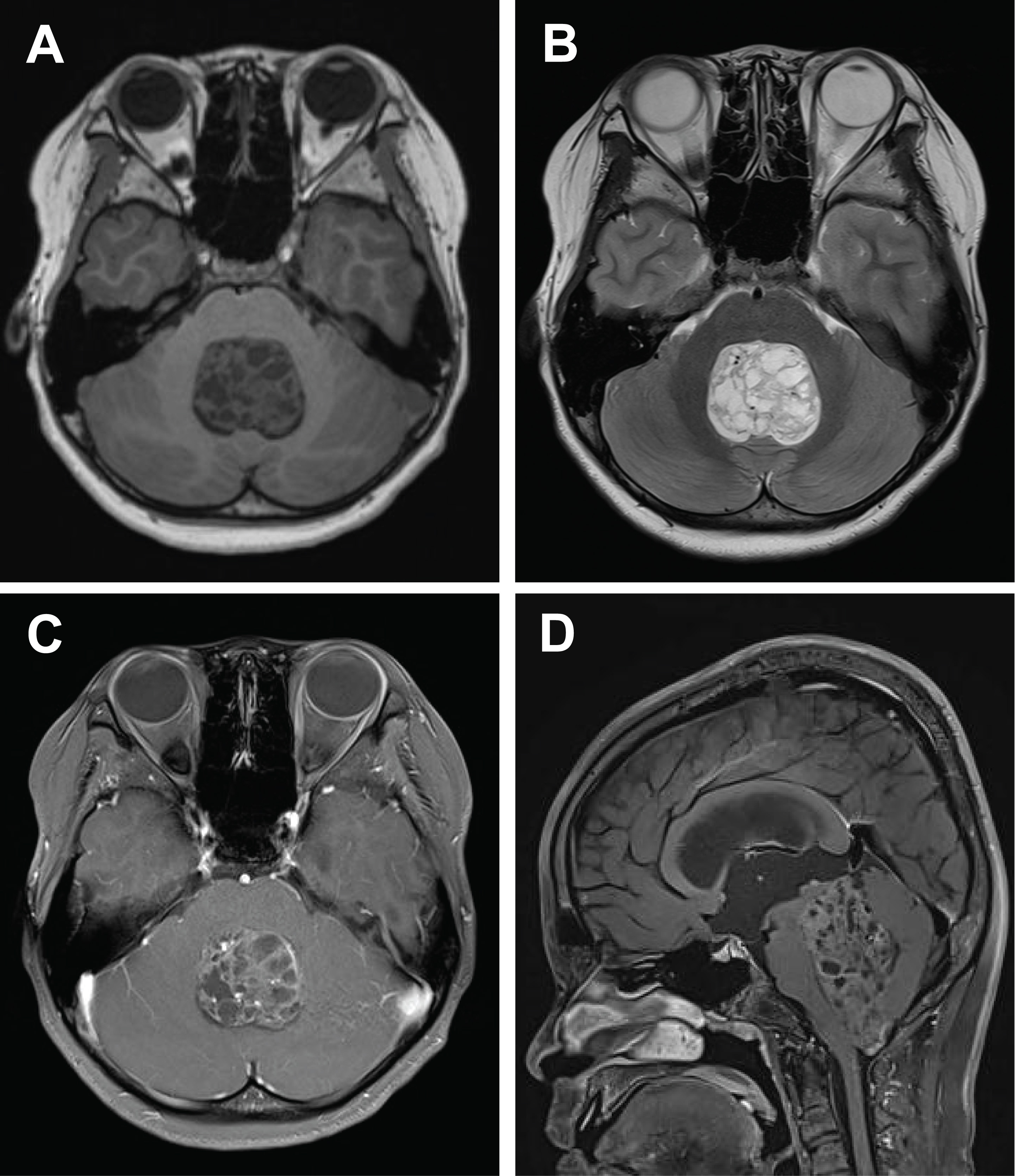
Preoperative magnetic resonance imaging in a 12-year-old boy presenting with progressive headaches, weight loss, and bilateral leg weakness over a 2-month period demonstrates a well-circumscribed 7.0 × 4.0 × 3.3 cm mixed solid and cystic mass located in the fourth ventricle. The lesion exhibits heterogeneous contrast enhancement with internal hemorrhage and calcification. (A) Axial T1-weighted image. (B) Axial T2-weighted image. (C) Gadolinium-enhanced axial T1-weighted image. (D) Gadolinium-enhanced sagittal T1-weighted image.
PATHOLOGIC FINDINGS
Macroscopically, these tumors demonstrate sharp circumscription from the surrounding brain parenchyma, although occasional cases may exhibit focal infiltrative borders. Histologically, the neoplasms are typically well-circumscribed but can display focal infiltration into adjacent brain tissue. They are characterized by a monomorphic population of round to oval or spindle-shaped cells with delicate chromatin and occasional glioma-like fibrillary processes or compact fascicular architecture. Architectural patterns demonstrate considerable variation, encompassing solid, microcystic, glioma-like, and hemangiopericytoma-like staghorn vasculature [15]. Some cases exhibit glial-like morphology with clear cytoplasm. A pathognomonic feature is the presence of ependymoma-like perivascular pseudorosettes (Fig. 2A–D). Myxoid or microcystic stroma and branching capillaries are frequently observed. Necrosis is commonly present, often accompanied by palisading, and mitotic activity is typically brisk. Microvascular proliferation is uncommon but has been documented focally in isolated cases [4,14].
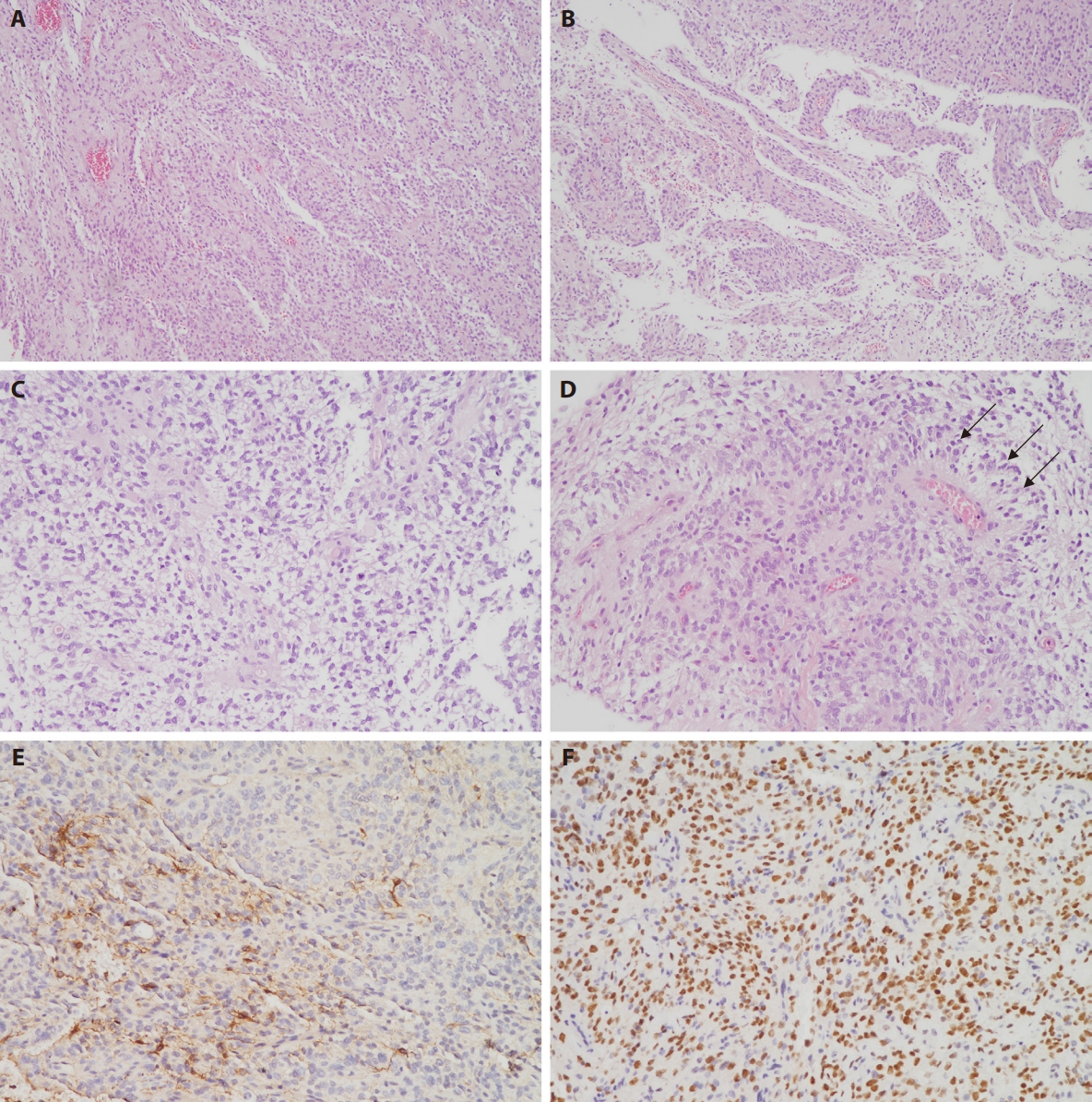
Histopathological features of the tumor resected from the fourth ventricle are demonstrated in Fig. 1. (A) Low-power photomicrograph demonstrates a solid growth pattern composed of monomorphic cells with focal slit-like spaces. (B) The tumor exhibits focal areas with papillary growth pattern. (C) Tumor components display delicate cytoplasmic processes with clear cell features. (D) Perivascular pseudorosettes are formed by delicate processes extending toward blood vessels (arrows). (E) Glial fibrillary acidic protein immunohistochemistry demonstrates positive staining in only a small subset of tumor cells. (F) BCL6 corepressor (BCOR) immunohistochemistry reveals strong nuclear positivity.
IMMUNOHISTOCHEMICAL PROFILE
Tumor cells consistently express vimentin and CD56, while demonstrating negative immunoreactivity for CD34, epithelial membrane antigen (EMA), desmin, and H3K27M. Variable and often focal expression of oligodendrocyte transcription factor 2 (OLIG2), glial fibrillary acidic protein (GFAP), and S100 has been documented (Fig. 2E), although diffuse GFAP positivity may occasionally occur [6]. NeuN expression is infrequently observed. Among 33 cases with available EMA immunohistochemistry results, positive expression was identified in seven cases (21.2%), including four cases with diffuse positivity [2], one case demonstrating a paranuclear dot-like pattern [13], one case with scattered expression [11], and our institutional case, which exhibited focal weak positivity. For synaptophysin, among 36 cases with reported results, 11 cases (30.5%) demonstrated focal positivity, including our institutional case, while all remaining cases were negative. Expression of H3K27me3 and ATRX is retained, while IDH1 R132H is negative. Ki-67 labeling index is elevated, ranging from 15% to 60%.
Nuclear BCOR immunoreactivity is widespread (Fig. 2F) but lacks specificity for BCOR ITDs. Among 48 previously reported cases with available BCOR immunohistochemistry results, only one demonstrated negative staining [5]. Diffuse nuclear positivity was observed in 42 cases (87.5%), and when including five additional cases with focal or weak-to-moderate staining, the overall positivity rate reaches 97.9%. However, nuclear BCOR immunoreactivity is not exclusive to ITDs and has been documented in tumors harboring BCOR gene fusions. In a prior cohort of 20 fusion-positive CNS tumors, 12 cases (60%) demonstrated BCOR positivity by immunohistochemistry, particularly those with EP300::BCOR and CREBBP::BCORL1 fusions [19]. Additionally, BCOR positivity has been reported in CNS tumors with KDM2A::YAP1 or KDM2B::NUTM2 fusions [5,20]. Notably, in a series of pineoblastomas, seven out of eight cases exhibited BCOR positivity despite lacking BCOR ITD or fusion events [21]. Other reports have described BCOR-positive tumors without any identifiable BCOR alterations [13], emphasizing the importance of cautious interpretation of BCOR immunohistochemistry results. SATB2 has been reported as a sensitive, though not specific, marker for BCOR-altered CNS tumors, with limitations due to intratumoral heterogeneity [22]. All previously reported cases (n = 10) subjected to SATB2 immunohistochemistry demonstrated positive nuclear expression.
MOLECULAR FEATURES
The pathognomonic diagnostic feature is a heterozygous ITD within exon 15 of the BCOR gene. DNA-based detection is generally sufficient to identify BCOR ITDs. However, DNA methylation profiling can assist in distinguishing BCOR ITD tumors from BCOR fusion-positive tumors, which exhibit distinct methylation signatures. Transcriptomic analysis is particularly useful for confirming BCOR fusions, especially when DNA-level alterations are inconclusive. Additional chromosomal abnormalities, including chromosomal gains and losses, may accompany BCOR ITDs [2].
ITD size demonstrates considerable variability, ranging from nine to 42 amino acids, with most duplications spanning approximately 20–30 residues [3]. A meta-analysis of 204 BCOR ITD–positive tumors revealed that the p.D1725_L1737 duplication is universally present, although the duplication size varies [16]. In our institutional case, a 13-amino-acid duplication was identified, representing the smallest reported standalone duplication to date. The protein sequences of the 44 cases in which the ITD sequence was identifiable are schematically illustrated in Fig. 3.

BCL6 corepressor (BCOR) protein sequence diagram illustrating internal tandem duplications (ITDs) in BCOR ITD–positive tumors. The frequency of each duplication pattern is expressed as percentages. Among the 68 previously reported cases and our institutional case, molecular protein sequences are available for 44 cases. Our institutional case demonstrates the shortest duplication size (asterisk).
DIFFERENTIAL DIAGNOSIS
Pediatric high-grade gliomas
Some tumors were initially misdiagnosed as anaplastic oligodendrogliomas due to their uniform cellularity, round nuclei with perinuclear halos, chicken-wire vasculature, and neuronal satellitosis [10]. Immunohistochemistry for H3K27M, H3K27me3, GFAP, OLIG2, S100, and BCOR helps distinguish these entities.
Glioblastoma
Several cases were initially misclassified as glioblastoma, particularly those lacking perivascular pseudorosettes and demonstrating highly cellular solid growth patterns [7,10]. In contrast to glioblastoma, BCOR ITD tumors typically lack prominent high-grade nuclear atypia, microvascular proliferation, and diffuse infiltration. Additionally, glial differentiation is difficult to confirm immunohistochemically in BCOR ITD tumors. BCOR and GFAP immunostaining provide valuable diagnostic assistance.
Embryonal tumor with multilayered rosettes
Similar to CNS tumors with BCOR ITD, embryonal tumor with multilayered rosettes (ETMR) predominantly affects children and demonstrates scant cytoplasm, spindle cells, and ovoid cells forming characteristic perivascular pseudorosettes. However, BCOR ITD tumors typically lack the well-defined perivascular anucleated zone characteristic of ETMR, and negative EMA expression in differential diagnosis.
Ependymoma
The presence of perivascular pseudorosettes necessitates careful differentiation from ependymoma. GFAP positivity within pseudorosettes is characteristic of ependymoma, whereas BCOR ITD tumors frequently demonstrate GFAP negativity. Loss of H3K27me3 expression in posterior fossa tumors can mimic posterior fossa group A ependymoma, making BCOR immunostaining crucial for accurate classification [9].
TREATMENT AND PROGNOSTIC FACTORS
Due to a limited number of reported cases, standardized treatment protocols and definitive prognostic factors remain poorly established. Treatment approaches have encompassed various chemotherapeutic regimens, including temozolomide monotherapy [7,15] and etoposide monotherapy [6]. However, multi-agent chemotherapy has been more commonly employed, utilizing combinations of temozolomide, etoposide, carboplatin, cisplatin, cyclophosphamide, vincristine, irinotecan, and bevacizumab [2,4,6-8,10-12,14-16,18].
No cases of cerebrospinal fluid dissemination have been reported at initial diagnosis. However, disease progression in some cases has included leptomeningeal dissemination [4], spinal metastasis [2,7], osseous metastasis [4], calvarial involvement [12], and subcutaneous spread [16,18].
Tumor recurrence is frequent, and overall survival remains poor [2]. Nevertheless, long-term survivors have been documented, including one case with survival exceeding 14 years following adjuvant chemoradiotherapy [10]. While cases with survival beyond 4 years exist, most patients ultimately succumb to the disease [6].
CONCLUSION
CNS tumors with BCOR ITD represent a rare but distinct pediatric neoplasm characterized by specific molecular and histopathological features. Although morphologic overlap with high-grade gliomas and ependymomas may present diagnostic challenges, an integrated approach incorporating histopathological assessment, immunohistochemical analysis, and molecular profiling enables accurate diagnosis. Future investigations are essential to establish optimal therapeutic strategies and elucidate the biological mechanisms underlying their aggressive clinical course.
Supplementary Information
The Data Supplement is available with this article at https://doi.org/10.4132/jptm.2025.07.23.
Notes
Ethics Statement
This study was approved by the Institutional Review Board of Chonnam National University Hwasun Hospital (CNUHH-2025-102) in accordance with the 1964 Helsinki declaration and its later amendments. Informed consent for publication was obtained from the patient's legal representative.
Availability of Data and Material
All data generated or analyzed during the study are included in this published article (and its supplementary information files).
Code Availability
Not applicable.
Author Contributions
Conceptualization: JHL, KHL. Data curation: JYL. Formal analysis: JYL, KSM. Funding acquisition: JHL, KHL. Investigation: SSK. Supervision: HJB, TYJ. Validation: HJB, TYJ, SSK. Visualization: JYL, KSM. Writing—original draft: JYL, KHL. Writing—review & editing: SSK, JHL. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
This work was supported by Chonnam National University Hospital Biomedical Research Institute (HCRI25039 for KHL).
Acknowledgments
The authors acknowledge the assistance of Claude Sonnet 4 (Anthropic, version 20250514) in manuscript preparation and English language editing.