Molecular Testing of Lung Cancers
Article information
Abstract
Targeted therapies guided by molecular diagnostics have become a standard treatment of lung cancer. Epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) rearrangements are currently used as the best predictive biomarkers for EGFR tyrosine kinase inhibitors and ALK inhibitors, respectively. Besides EGFR and ALK, the list of druggable genetic alterations has been growing, including ROS1 rearrangements, RET rearrangements, and MET alterations. In this situation, pathologists should carefully manage clinical samples for molecular testing and should do their best to quickly and accurately identify patients who will benefit from precision therapeutics. Here, we grouped molecular biomarkers of lung cancers into three categories—mutations, gene rearrangements, and amplifications—and propose expanded guidelines on molecular testing of lung cancers.
Molecular diagnostics-guided targeted therapies have become a standard treatment for patients with lung cancer [1]. Driver genetic alterations such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) rearrangements are currently used as predictive biomarkers for EGFR tyrosine kinase inhibitors (TKIs) and ALK inhibitors, respectively [1]. Since Korean guideline recommendations for EGFR and ALK molecular testing were published [2,3], the list of druggable genetic alterations has been growing as tremendous amounts of information on the cancer genome are becoming available (Table 1) [4-6]. Molecular analyses of various biomarkers in tumor tissue or cytology specimens have become standard laboratory tests for the clinical management of lung cancers. In this context, an updated and more comprehensive set of guidelines is necessary.
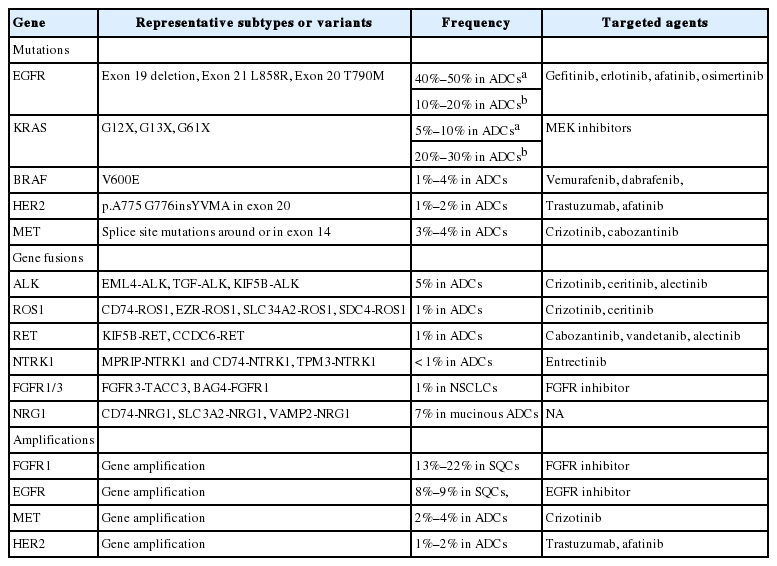
Targetable genetic alterations in lung cancer
In this article, we propose expanded guidelines for molecular testing of lung cancers including recently updated genetic alterations and well-known biomarkers. Here, we grouped molecular biomarkers into three categories—mutations, gene rearrangements, and amplifications (Fig. 1)—and reviewed the background, indications, methods, reporting, test validations, and quality assurance for each category.
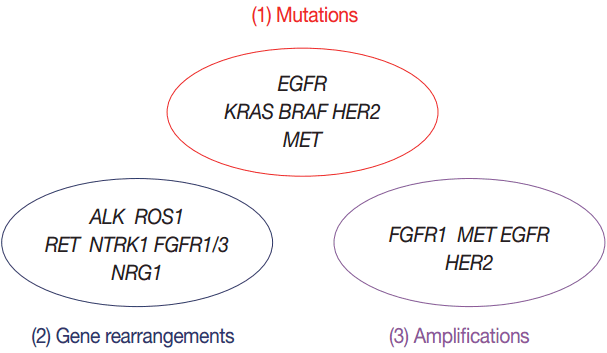
Three categories of molecular biomarkers used in these guidelines. EGFR, epidermal growth factor receptor; ALK, anaplastic lymphoma kinase; FGFR, fibroblast growth factor receptor.
MOLECULAR TESTING
Mutations including EGFR, KRAS, BRAF, HER2, and MET
Background
Activating somatic mutations including point substitution, small insertion, and in-frame deletion are major oncogenic drivers in lung cancer. The discovery of activating EGFR mutations and their close relation with the response to EGFR TKIs opened the new era of precision medicine. Since then, clinical trials have confirmed that EGFR mutations are the best predictive factor of EGFR TKI efficacy [7,8]. EGFR mutations have been more frequently found in lung adenocarcinomas in Asian populations than Western populations [9]. The overall EGFR mutation rate is approximately up to 46% among Korean patients with adenocarcinoma [10,11]. More than 90% of sensitizing EGFR mutations are composed of an in-frame deletion in exon 19 and a point mutation (L858R) in exon 21. Nonetheless, most patients experience disease progression usually after 12 months of treatment [12,13]. A variety of mechanisms are involved in acquired resistance to EGFR TKIs [12-14]. Among these, the most common resistance mechanism is the T790M mutation in EGFR, constituting 50%–60% of cases [12,13,15]. Recently, osimertinib was approved for the treatment of T790M-positive lung cancers in Korea [16]. The acquired EGFR C797S mutation has been reported to mediate resistance to osimertinib in a subset of EGFR T790M–positive non-small cell lung carcinomas (NSCLCs) [17,18]. Thus, EGFR mutation testing covering sensitizing as well as nonsensitizing mutations has become the most important step in treatment decision-making for lung cancer patients because of the high frequency of EGFR mutations and the availability of targeted therapeutic agents.
The frequency of KRAS mutations has been reported to be 8%–15% in lung adenocarcinoma among Korean patients [19,20]. Unfortunately, targeted therapies thus far have been unsuccessful [21]. KRAS mutations are known as negative predictive markers of a poor response to EGFR TKIs. In addition, a KRAS mutation has been reported to be a poor prognostic factor. This also appears to be dependent both on the specific KRAS codon mutation and disease stage at the time of diagnosis [21]. Invasive mucinous adenocarcinomas show a high incidence of KRAS mutations [22].
Besides EGFR mutations, other targetable mutations have been found although their frequency is low. A BRAF mutation has been found in 3%–4% of lung adenocarcinomas in Western patients [23,24]. Among never-smoker Korean women with lung cancer, the frequency of tumor BRAF mutation is 1% [25]. The V600E mutation constitutes only 50% of BRAF mutations in contrast to other cancers (such as melanoma and papillary thyroid carcinoma) [23,24]. Vemurafenib and dabrafenib show clinical activity in BRAF V600E-mutant NSCLC [26-28]. HER2 mutations are present in approximately 1%–2% of lung adenocarcinomas [29]. The most common mutation is an in-frame insertion within exon 20 [29]. The exon 20 insertion results in increased HER2 kinase activity. The clinical response to HER2-targeting agents, such as trastuzumab and afatinib, is observed in patients with lung cancer harboring a HER2 mutation [29].
Recently, MET splice site mutations have emerged as targetable oncogenic drivers. Oncogenic mutations in the MET exon 14 splice sites (these mutations cause exon 14 skipping) occur in 3%–4% of lung adenocarcinomas [30,31]. Patients with lung cancer harboring MET exon 14 skipping show a clinical response to MET inhibitors including crizotinib [30,31].
Indications
The important reason for molecular testing of lung cancers is to select patients who may benefit from targeted therapies. In addition, patients with lung cancer can get benefits from molecular testing of their tumors regardless of stage. For example, molecular testing can provide accurate information on staging (in case of multiple tumors), prognostic stratification, and prompt treatment in case of recurrence. Because of the high frequency of EGFR mutations in Asian populations as mentioned above, EGFR mutation testing is especially important for the treatment of Korean lung cancer patients. The Korean Food and Drug Administration (FDA) approved only EGFR molecular testing from among five genes important for decision-making with respect to first-line chemotherapy during standard treatment. Other mutations can also be approved and used as predictive markers in the near future. Each mutation is significantly associated with some clinical factors or histological subtypes. Nevertheless, clinical findings alone cannot completely predict specific mutation status [32,33]. In most of the guidelines published so far, histological types have been recommended as the most important factor in determining whether to perform molecular tests [34-36]. In particular, for patients who have a diagnosis of NSCLC with an adenocarcinoma component or nonsquamous cell type, molecular testing is routinely recommended [35]. Thus, pathologists should try to further classify NSCLCs into more specific subtypes, such as adenocarcinoma or squamous cell carcinoma (SQC) [2]. In addition, a minimum immunohistochemical panel, such as one adenocarcinoma marker and one SQC marker, is recommended to preserve as much tissue as possible for molecular testing in small tissue samples [35,37]. Besides histological analysis, in cases of young age, female gender, never-smokers, small biopsies, or patients with a combined tumor type, molecular testing can be done.
Methodology
Methods
A variety of methods can be used for detecting mutations including direct sequencing, real-time polymerase chain reaction (PCR), and commercial kits (Table 2) [38,39]. Pathologists should consider the available approaches and the advantages and disadvantages of each method, including analytical sensitivity and turnaround time. In addition, new technologies for molecular testing must be approved by the Korean government before incorporation into clinical practice.
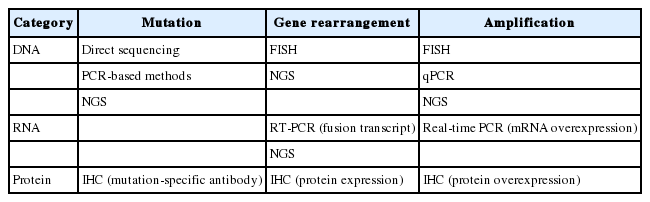
Representative methods categorized by mechanisms of oncogene activation and by targeted molecules
Direct sequencing is considered the gold standard for mutation analysis. On the other hand, direct DNA sequencing requires a high ratio of tumor tissue to normal tissue (more than 50% tumor content) for reliable results. In contrast, PCR-based methods show high sensitivity, requiring mutant DNA content of only 1% [38]. Nevertheless, these PCR-based methods can detect only previously known mutations or targeted sites. PCR-based methods including the peptide nucleic acid–mediated PCR clamping method and allele-specific PCR method have been approved in Korea. Among the targetable mutations, EGFR mutations have been the most studied in the field of lung cancer. In studies comparing two representative methods, clinical outcomes are not significantly different among groups harboring EGFR mutations detected by direct sequencing or by PCR-based methods [40,41]. Highly sensitive methods may also be useful for the detection of EGFR mutations associated with acquired resistance, e.g., T790M [15]. As with the EGFR mutation, direct sequencing or real-time PCR can be used for the detection of KRAS, BRAF, and HER2 mutations, which include a point mutation or small indel. In the case of MET exon 14 alterations, most alterations are a point mutation or indel, affecting the splice acceptor site, splice donor site, or the approximately 25-base pair (bp) intronic noncoding region. In rare cases, however, whole-exon deletions of MET exon 14 have been reported [42]. A DNA-based method including direct sequencing is possible, but the mRNA-based method including real-time PCR is better at detecting MET exon 14 skipping.
As targetable genetic alterations are increasingly discovered, individual genotyping may become relatively inefficient and costly. Thus, next-generation sequencing (NGS) technology with DNA or RNA is reported to be useful for multiplexed and deep genomic sequencing [43,44]. Especially, targeted deep sequencing of selected gene sets (a so-called cancer panel) is approaching integration into daily clinical practice. Nonetheless, due to the methodological complexity of NGS, analytical validation and quality control are crucial for implementation in routine practice. Furthermore, to become a companion diagnostic assay, an NGS-based method should be comparatively validated with previous companion diagnostics.
Sample types
As previously described in our proposed guideline, small biopsy and cytology samples are used for mutation testing [2]. In particular, all samples obtained from a variety of methods are acceptable, including transbronchial lung biopsy, endobronchial ultrasound-guided transbronchial needle aspiration, bronchial brushing or washing, computed tomography–guided gun biopsy or needle aspiration, and pleural fluid sampling [34,45,46]. The results using cytology samples are highly concordant with those of the corresponding tissue samples, especially with more sensitive methods [45,47].
Since most driver mutations develop in early steps of carcinogenesis, tumor samples from either primary masses or metastatic lesions are equally suitable for mutation testing [48]. In the case of multiple primary lung cancers, each tumor may be tested [49].
Formalin-fixed, paraffin-embedded (FFPE) tissues are most frequently used in molecular testing. Routinely prepared FFPE tissues are the most practical resource for molecular analysis, in spite of fixation-related artifacts [50]. The optimal fixative for preparing FFPE samples is generally 10% neutral-buffered formalin [34]. The optimal fixation time ranges from 6 to 72 hours to avoid underfixation or overfixation, respectively [34,50]. Routinely prepared cytology samples such as alcohol-fixed smears or Thin Prep slides [45,47] and cell block samples [51] are also suitable for mutation testing.
Because obtaining a tissue sample from bronchoscopy or a transthoracic needle biopsy is difficult and nonrepeatable, a liquid biopsy, which contains circulating tumor DNA, circulating tumor cells, or exosomes using plasma or body fluids, has recently been attracting attention as a new source for identification of somatic mutations [52,53]. Nonetheless, due to the high rate of false negative results at present, negative results require a tissue biopsy to determine exact mutation status [53].
Sample requirements
Pathologists must confirm the presence of tumor cells in a sample before mutation analysis. The percentage and quality of tumor cells are crucial for proper mutation testing. For example, direct sequencing requires at least 20% tumor cells in the sample for reliable testing result. Thus, evaluation of the percentage of tumor cells (tumor purity) in a given sample is highly recommended, especially for less sensitive methods. Tumor dissection may be used to increase the tumor content if required. Thus, as noted, the minimum number or percentage of cancer cells required for adequate testing depends on the analytic sensitivity of the testing method.
Reporting
Mutation testing reports should contain identification of the patient (pathology number, hospital unit number, age, and gender), requesting physician with department, receipt day, report day, sample used for the testing (site or organ, sample type, and tumor purity), methodology used, exons tested, test results, comments on results, and names of the testing technician and corresponding pathologist [2].
Validation of a test
Analytical and clinical validation should be done when the testing is implemented in the laboratory. One of the references is indexes of evaluation from the Korean Institute of Genomic Testing Evaluation.
Repeat examination can be considered in cases of poor sequencing data, results not matching previously well-defined clinical features or a unique histological subtype, or concurrent detection of mutually exclusive driver mutations. Other types of methods may be useful in equivocal cases.
Quality assurance
For optimal mutation testing, the quality of a sample and validation status of the testing method are crucial. Laboratories must incorporate mutation testing methods into their overall laboratory quality improvement program, by establishing appropriate quality improvement monitors as needed to ensure consistent performance at all steps of the testing and reporting processes. In particular, laboratories should participate in a formal proficiency testing program, if available, or an alternative proficiency assurance activity. There is an external quality control program for mutation testing from the Korean Institute of Genomic Testing Evaluation and the Korean Society of Pathologists. To improve the reliability of assays in terms of detection of mutations, a regular quality control program should be implemented, such as examination of reference materials or comparison of the results between different diagnostic assays.
Gene rearrangements including ALK, ROS1, RET, NTRK1, NRG1, and FGFR
Background
Since the discovery of a transforming fusion gene between echinoderm microtubule-associated protein-like 4 (EML4) and ALK in 2007, the list of targetable gene fusions in lung cancer has been growing [54,55]. Although the prevalence of each gene fusion is low (approximately 1%–5%), molecularly tailored treatments have resulted in dramatic clinical responses. The presence of an ALK rearrangement and ROS1 proto-oncogene receptor tyrosine kinase (ROS1) rearrangement in lung cancer has become the best predictor of response to crizotinib [56,57]. Subsequently, RET proto-oncogene (RET) fusions were found comprising 1% of lung adenocarcinomas [58,59]. Cabozantinib or vandetanib showed clinical activity against lung cancer harboring RET fusions [60,61]. Besides ALK, ROS1, and RET fusions, NTRK1 and fibroblast growth factor receptor (FGFR) 1/3 fusions were reported in lung cancer, which are expected to give a therapeutic opportunity to patients with no known driver alterations [62,63]. Of note, NRG1 fusions were predominantly found in invasive mucinous adenocarcinoma, which is a unique variant of lung adenocarcinoma [22,64].
Indications
Clinical characteristics associated with gene fusions such as ALK, ROS1, RET, and NTRK1 are adenocarcinoma histological features, never- or light-smoking history, and younger age. Nevertheless, not all gene fusions have these characteristics. FGFR fusions are associated with smoking history [63]. Therefore, clinical characteristics alone cannot determine gene rearrangement testing.
Histological type is an important factor in determining whether to perform genetic analysis or which genes to test [36]. Most targetable gene fusions are discovered in NSCLCs. Because ALK, ROS1, and RET fusions are predominantly found in adenocarcinomas or NSCLCs with an adenocarcinoma component, pathologists should try to further classify NSCLCs into more specific subtypes. On the other hand, FGFR fusions can be found in adenocarcinomas and SQCs [63,65].
Methodology
Methods
Several methods are currently available for assessing gene rearrangement, including fluorescence in situ hybridization (FISH), immunohistochemistry (IHC), reverse-transcriptase polymerase chain reaction (RT-PCR), and NGS (Table 2).
Break apart FISH is the currently standard method for the detection of gene rearrangements. Although the FISH assay requires technical expertise and experience for interpretation, this assay is reasonably sensitive and specific for the detection of gene rearrangements regardless of fusion partners. The pathologist should recognize the probe design, especially which sides are labeled with a red and green signal and the positive criteria. Crizotinib, a first generation ALK inhibitor, was approved by the United States FDA in conjunction with a companion diagnostic FISH test (ALK Break Apart FISH Probe Kit, Abbott Molecular Inc.). Positive criteria for the Abbott FISH probe kit are as follows. Positive signals are defined as separate signals or a single red signal. Initially, 50 tumor cells are counted. A sample is considered positive if > 25 cells out of 50 (> 25/50 or > 50%) test positive. A sample is considered equivocal if 5 to 25 cells (10 to 50%) test positive. If the sample is equivocal, a second reader should evaluate the slide. The first and second cell count readings are added up, and a percentage is calculated out of 100 cells. If the percentage of positive cells is ≥ 15% (≥ 15/100), the sample is considered positive. The Korean FDA also approved the ALK FISH as a selection test for crizotinib treatment in lung cancer patients.
Because protein expression can serve as a surrogate marker of gene rearrangements, IHC is useful for screening of gene rearrangements when it is appropriately validated [3,66-68]. In particular, ALK IHC was extensively studied and validated [3]. Among ALK IHC antibodies, 5A4 and D5F3 appear to be superior to the ALK1 antibody for detecting ALK-rearranged lung cancer [69,70]. IHC can effectively predict ALK fusion status because lung adenocarcinoma in general does not express the ALK protein without genetic alterations. The VENTANA ALK (D5F3) CDx Assay was approved by the U.S. FDA for identification of patients eligible for treatment with crizotinib. Other antibodies are not validated as companion diagnostics; therefore, their use should be limited to a screening step within controlled settings.
Besides ALK, there are no approved companion diagnostics for other gene rearrangements. Break apart FISH is generally used to detect gene arrangements in clinical samples, and IHC can serve as a screening method.
In terms of mRNA expression, the RT-PCR assay is used to find fusion transcripts with unique sequences. RT-PCR methods are generally known to be sensitive [71]. Nonetheless, RT-PCR has some limitations in clinical practice. Primer design requires information about fusion partners and breakpoints. Thus, only known fusion variants can be detected. In addition, most materials for molecular testing are FFPE samples, where RNAs can be severely degraded compared to fresh frozen tissue. Nevertheless, RT-PCR can be useful when only cytology samples are available [71].
The NGS technology has emerged as an alternative to the detection of gene rearrangements. It can simultaneously detect a variety of gene fusions using targeted DNA sequencing or RNA sequencing [43,44]. The sensitivity and specificity for calling a gene fusion mainly depend on the detection algorithm. The NGS-based gene rearrangement method should be vigorously validated before implementation in routine practice. Furthermore, to become a companion diagnostic test, an NGS-based method should be comparatively validated with previous companion diagnostics.
Sample type
A variety of biopsy specimens can be used for gene rearrangement testing as described above in the section on mutation testing. Previous studies have shown that cytology samples are suitable for gene rearrangement analysis, especially as validated in ALK fusion testing [72]. The cell block is usually first used if there are enough tumor cells in the block. Smear slides can also be used for the detection of gene rearrangement [72]. Thus, both tissue and cytology samples are acceptable for gene rearrangement testing, if appropriately validated. Details were described in the previous guideline [3].
Sample requirements
For adequate evaluation of break apart FISH, a minimum of 50 to 100 well-preserved tumor cells are required. Tumor purity is not as critical for FISH tests, however, it is necessary to choose areas of a slide in which the viable tumor cells are most abundant. Because a dark-field fluorescence microscope does not provide perfect visualization of tumor morphology, the pathologist should determine whether the selected area contains a sufficient number of tumor cells and should interppret the result carefully by distinguishing between cancer cells and intimately mixed non-neoplastic cells.
As for RT-PCR and NGS technologies, the tumor percentage can be critical in gene rearrangement testing as described in mutation testing. The pathologist should verify the quality of the sample and the presence of tumor cells. Tumor dissection may be used to increase the tumor content if required.
Reporting
Reports on gene rearrangement testing should contain identification of the patient (pathology number, hospital unit number, age, and gender), requesting physician with department, receipt day, report day, sample used for the testing (site or organ, sample type, and tumor purity), methodology used, detectable fusion variants, test results (negativity or positivity for the gene rearrangement), comments on results, and names of the testing technician and corresponding pathologist. We recommend reporting the additional details in the case of a FISH test: information on probe design, total number of counted nuclei, and percentage of the tumor nuclei showing positive signals (split signal or isolated signal containing a kinase domain) [3].
Validation of the test
General principles are similar to those of mutation testing. Technical and clinical validation procedures should be carried out when the testing is set up in the laboratory. One of the references is indexes of the evaluation from the Korean Institute of Genomic Testing Evaluation.
Pathologists should recognize the limitations of each methodology. With respect to the break apart FISH test, repeat examination can be considered in cases of poor signals, atypical signal patterns (including predominantly isolated 5' signals), a borderline range that approached the 15% cutoff line, results not matching well-validated IHC, or concurrent detection of driver mutations. Other types of methods may be useful in equivocal cases.
Quality assurance
There is an external quality control program for ALK fusion testing from the Korean Institute of Genomic Testing Evaluation and the Korean Society of Pathologists. To improve the reliability of assays for detecting gene rearrangements, a regular quality control program should be performed, and the results of molecular testing can be confirmed by different diagnostic assays, such as validated IHC.
Amplifications including FGFR1, EGFR, MET, and HER2
Background
Gene amplification is an important mechanism for oncogene activation. HER2-targeting therapy has been implemented in breast and gastric cancer on the basis of molecular testing for HER2. Amplifications of several genes have been studied in lung cancer.
FGFR1 is one of the most commonly amplified genes in human cancers. Amplified FGFR1 has been described in 13%–22% of lung SQCs [73,74]. It is expected to become a promising targetable alteration in lung SQCs because there are no known targeted therapeutic agents for SQCs [73]. In vivo studies have revealed that inhibition of the FGFR1 pathway with FGFR inhibitors leads to significant tumor shrinkage, suggesting that FGFR inhibitors might be an effective therapeutic option in SQCs with FGFR1 amplification [75]. Nevertheless, clinical trials of FGFR inhibitors were not impressive [76]. Additional biomarkers should be studied to predict the drug response to FGFR inhibitors [77]. A study showed that homogeneous high-level amplification was associated more strongly with a clinical response to FGFR inhibition [78]. Thus, FGFR1 amplification can still be a promising candidate for targeted therapy.
EGFR amplifications were found in adenocarcinomas and SQCs. Initially, EGFR amplification was regarded as a predictive biomarker of EGFR TKI treatment [79]. Subsequent studies revealed that EGFR amplification was closely related to EGFR mutation, and EGFR mutation was the strongest predictive marker of the response to EGFR TKIs [7,80]. Among SQCs, EGFR high copy number gains/amplifications were present in 8%–9% of cases [4,81]. Patients with lung SQC harboring an EGFR high copy number gains or amplifications showed a higher response rate to EGFR TKI [81].
De novo MET amplification was present in 2%–4% of lung adenocarcinomas [5,82]. NSCLC patients with de novo MET amplification showed a rapid and durable response to crizotinib [83,84]. MET amplification was also identified as one of the acquired secondary resistance mechanisms in patients with EGFR mutations who progressed on EGFR TKIs [12,13].
De novo HER2 amplification was found to be present in 1%–2% of lung adenocarcinomas [5]. NSCLC patients with de novo HER2 amplification can be treated with HER2-targeting agents [85]. HER2 amplification was also identified as one of the acquired secondary resistance mechanisms in patients with EGFR mutations who progress on EGFR TKIs [18,86].
Indications
Amplifications of receptor tyrosine kinase genes are present in lung cancers regardless of histological subtypes, while the frequencies are different. Thus, these gene amplifications can be examined in patients with lung cancer, especially previously targetable oncogene-negative cases. FGFR1 amplification is especially meaningful for SQCs.
EGFR amplification can serve as a predictive marker of SQCs. MET and HER2 amplification can be examined in the primary tumors or tumors with acquired resistance to TKIs.
Methodology
Method
Several methods can be used for detection of gene amplifications (Table 2). FISH using a locus-specific intensifier (LSI) gene and a chromosome-specific centromere (CEP) probe is a standard method for the detection of gene amplifications. Although the FISH assay requires technical expertise and experience for interpretation, this method has the advantage of being able to evaluate gene amplification by selecting only cancer cells.
FISH-positive criteria have been used according to the corresponding genes. FGFR1 high-level amplification is defined as copy number ≥ 9 or (1) an FGFR1/CEP8 ratio of ≥ 2.0, (2) the average number of FGFR1 signals per tumor cell nucleus ≥ 6, and (3) the percentage of tumor cells containing ≥ 15 FGFR1 signals or large clusters ≥ 10% [74,87]. According to previously published criteria, the EGFR gene copy number was classified into six FISH strata: disomy (two or fewer copies in more than 90% of cells), low trisomy (two or fewer copies in 40% or more of the cells, three copies in 10%–40% of cells, and four or more copies in less than 10% of cells), high trisomy (two or fewer copies in ≥ 40% of cells, three copies in ≥ 40% of cells, and four or more copies in less than 10% of cells), low polysomy (four or more copies in 10%–40% of cells), high polysomy (four or more copies in 40% of the cells or more), and gene amplification (defined by the presence of tight EGFR clusters and a ratio of EGFR to the chromosome of ≥ 2.0, or ≥ 15 copies of EGFR per cell in ≥ 10% of the cells analyzed) [79]. Regarding MET, FISH-positive groups include (1) high-level amplification (presence of loose or tight clusters of MET signals too numerous to count) or a MET/CEP7 ratio greater than 5.0 and (2) low-level amplification (tumors with the MET/CEP7 ratio ≥ 2.2 and ≤ 5.0) [83]. As for HER2, gene amplification can be defined as positive when the HER2/CEP17 ratio is ≥ 2.0 or average HER2 copy number is ≥ 6.0 signals/cell based on the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines for dual-probe in situ hybridization [88].
In clinical practice, for many years FISH has been the most widely used method for gene copy number assessment. Silver in situ hybridization (SISH) is a new technology with some clinical advantages over FISH. Examination can be made using conventional light microscopy with preserved tissue architecture. In addition, the slides are durable and can be reviewed several years after staining. Thus, the SISH technique can be more practically applied in routine diagnostic procedures [89].
Quantitative real-time PCR can be used to detect gene amplification [90]. In this case, the optimal cutoff threshold of amplification should be determined considering clinical relevance and a response to targeted therapeutic agents.
The NGS technology can simultaneously detect a variety of genes with a copy number gain using DNA sequencing [43]. Nonetheless, currently, the NGS algorithm for determination of a copy number gain in heterogeneous tumor tissue is not well established. The established guideline is based on a specific gene copy number, but the NGS technology cannot determine the specific gene number in given cancer cells. The reliability of testing results also depends on the tumor cell percentage and the degree of gene amplification. Before clinical implementation, thorough verification is required.
Sample type
Various biopsy samples can be used for amplification testing as in other molecular testing methods described above. Samples routinely consist of FFPE tissues. Tumor tissues from either primary tumors or metastatic lesions can be equally available for molecular testing especially regarding high-level homogeneous amplification [91]. Cytology samples can be used with quantitative real-time PCR or NGS.
Sample requirements
Regarding the in situ hybridization method, a minimum of 50 to 100 tumor cells are required. General principles are similar to those described above in the section on break apart FISH testing.
Reporting
Reports on gene amplification testing should contain identification of the patient (pathology number, hospital unit number, age, and gender), requesting physician with department, receipt day, report day, sample used for the testing (site or organ, sample type, and tumor purity), methodology used, test results (negativity or positivity for the gene amplification according to proposed criteria), comments on results, and names of the testing technician and corresponding pathologist. Additional information can be included in the case of in situ hybridization: the total number of counted nuclei, gene/CEP ratio, average copy number, or findings according to special diagnostic criteria.
Validation of a test
Analytical and clinical validation procedures should be conducted when the testing is set up in the laboratory. One of the references is indexes of evaluation from the Korean Institute of Genomic Testing Evaluation. General principles are similar to the ASCO/CAP guidelines for dual-probe in situ hybridization [88].
Quality assurance
There is an external quality control program for FISH/SISH testing (for example, HER2 amplification) from the Korean Institute of Genomic Testing Evaluation and the Korean Society of Pathologists. To improve the reliability of assays for detection of gene amplification, a regular quality control program should be implemented, and the results of molecular testing can be confirmed by different diagnostic assays, such as validated IHC for protein overexpression.
CONCLUSION
We proposed guideline recommendations for molecular testing according to three categories of genetic alterations in lung cancer (summarized in Table 3). Molecular diagnostics for targetable genetic alterations have become the standard of care in the management of lung cancer patients (Fig. 2). In addition, immunotherapy has emerged as a new therapeutic option for patients who do not have targetable alterations (Fig. 2). Both therapeutic approaches require pathological diagnosis based on molecular biomarker assessment. Therefore, pathologists should carefully manage clinical samples for molecular testing and should do their best to quickly and accurately identify patients who will benefit from precision therapeutics. Each pathology department should fully validate the detection methods and should also participate in a quality control and formal proficiency testing program.
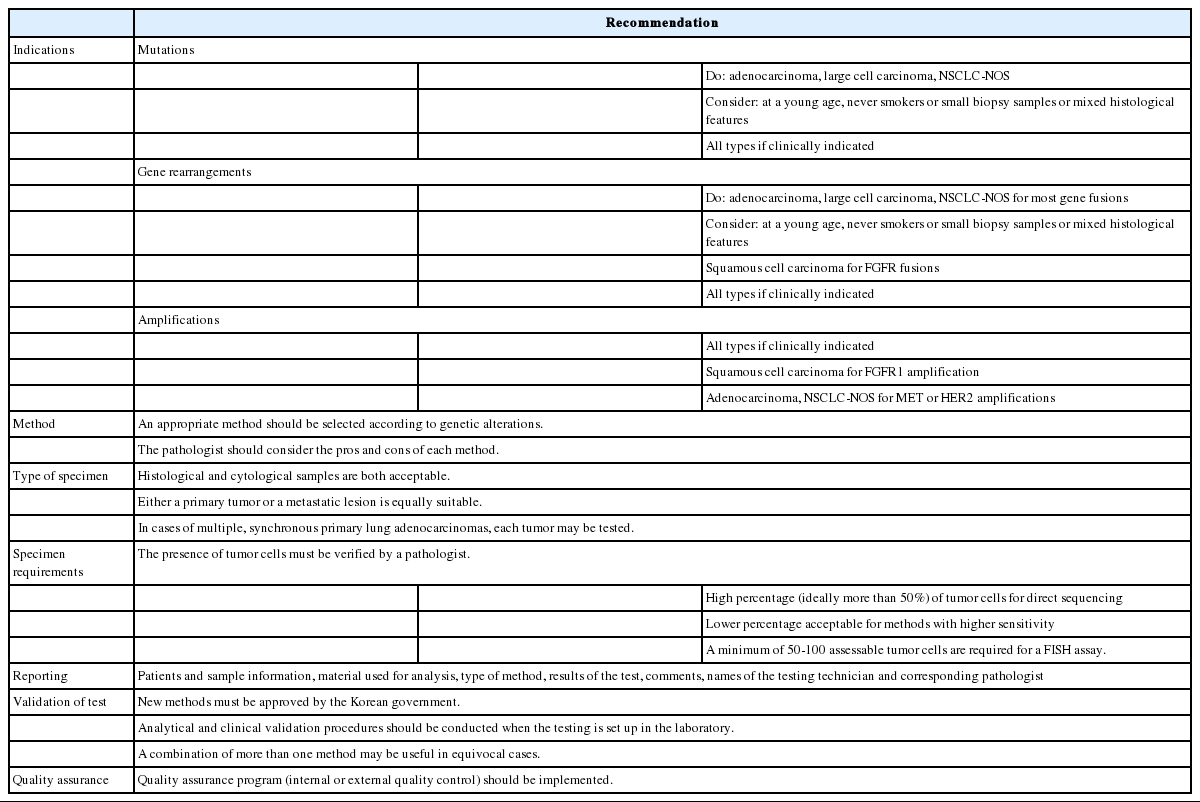
A summary of recommendations for molecular testing of lung cancer
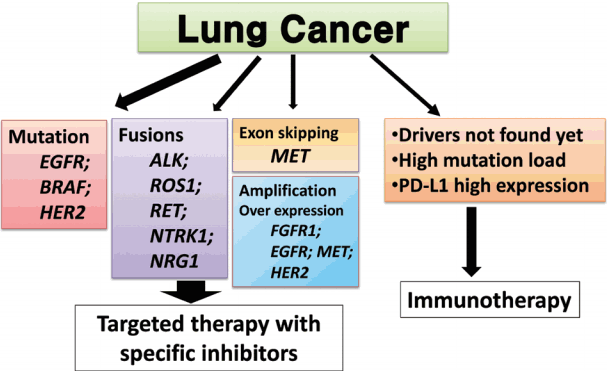
The current therapeutic approach to patients with lung cancer. EGFR, epidermal growth factor receptor; ALK, anaplastic lymphoma kinase; FGFR1, fibroblast growth factor receptor 1; PD-L1, programmed death-ligand 1.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by The Korean Society of Pathologists Grant (2016).