Inflammatory bowel disease–associated intestinal fibrosis
Article information

Abstract
Fibrosis is characterized by a proliferation of fibroblasts and excessive extracellular matrix following chronic inflammation, and this replacement of organ tissue with fibrotic tissue causes a loss of function. Inflammatory bowel disease (IBD) is a chronic inflammation of the gastrointestinal tract, and intestinal fibrosis is common in IBD patients, resulting in several complications that require surgery, such as a stricture or penetration. This review describes the pathogenesis and various factors involved in intestinal fibrosis in IBD, including cytokines, growth factors, epithelial-mesenchymal and endothelial-mesenchymal transitions, and gut microbiota. Furthermore, histopathologic findings and scoring systems used for stenosis in IBD are discussed, and differences in the fibrosis patterns of ulcerative colitis and Crohn’s disease are compared. Biomarkers and therapeutic agents targeting intestinal fibrosis are briefly mentioned at the end.
Fibrosis is characterized by fibroblastic proliferation and the deposition of excessive extracellular matrix (ECM) and hard collagen layers through chronic inflammatory and reparative responses [1–4]. Fibrosis can also occur during the wound healing process, and it is an end-stage phenomenon that causes chronic damage to solid organs, such as the liver, lungs, and kidneys [5]. Hepatitis and cholestatic diseases with various etiologies eventually lead to liver cirrhosis, and various types of glomerulonephritis lead to glomerulosclerosis. In such cases, the organ is replaced with fibrous tissue instead of the original functional tissue, resulting in a loss of function. However, in contrast to those solid organs, the phenomenon of fibrosis in the gastrointestinal tract, a hollow viscous organ, is poorly understood.
Inflammatory bowel disease (IBD) is characterized by persistent and refractory immuno-mediated inflammation in the gastrointestinal tract [6]. To date, studies on disease activity in IBD have focused on the presence or absence of ulcers and the degree of infiltration of inflammatory cells. However, intestinal fibrosis is a common pathogenic feature of IBD and has several related complications, including stricture, fistula, and bowel penetration [7,8]. Intestinal fibrosis can occur through several etiologies, including a desmoplastic reaction, radiation enteropathy, solitary rectal ulcer, graft-versus-host disease, post-surgical adhesions, desmoid tumors, collagenous colitis, and eosinophilic enteropathy. IBD is the leading cause of intestinal fibrosis, and it is thus necessary to understand its unique form of intestinal fibrosis [9–11].
Intestinal fibrosis is a pathological complication of great interest not only to radiologists and pathologists but also to clinicians due to its association with the risk of major complications that require surgical treatment, prognosis after surgery, and the risk of recurrence [12].
PATHOGENESIS
Fibrosis is an irreversible process that occurs as a consequence of chronic inflammation. It results in persistent luminal narrowing and strictures. Anti-inflammatory agents do not prevent or treat fibrosis in IBD, even if they improve the inflammation [13]. Because the fibrotic process is not affected by various IBD treatments, researchers have focused on inflammation-independent mechanisms, such as genetic factors, environmental risks, and the gut microbiota, which are known to affect the prognosis of fibrosis [14]. Moreover, intestinal fibrosis can be observed alongside excessive deposition of ECM and activated mesenchymal cells in the intestinal wall [15]. The main known drivers of this fibrosis mechanism are soluble molecules (cytokines and growth factors), the epithelial-mesenchymal transition (EMT), the endothelial-mesenchymal transition (EnMT), and the gut microbiota.
Genetic factors
Genetic research on IBD has proposed several genetic pathways for its pathogenesis [16,17]. However, relations between those gene mutations and intestinal fibrosis have not yet been well studied. In one bioinformatics study, researchers hypothesized that similar molecular pathways would be involved in fibrosis in various organs and thus measured gene expressions found in kidney fibrosis and liver cirrhosis in Crohn’s disease (CD) and ulcerative colitis (UC). They found that fibrosis in different organs had different gene signatures. C-X-C motif chemokine ligand 9 (CXCL9) and CD52 were upregulated in both CD and UC, whereas thrombospondin 2 (THBS2), matrix gla protein (MGP), protein tyrosine phosphatase receptor type C (PTPRC), and decorin (DCN) were upregulated only in CD. In UC, CXCL9, CD52, and granzyme A (GZMA) were upregulated, and DCN, which was elevated in CD, was downregulated [18].
Nucleotide binding oligomerization domain containing 2 (NOD2) has been most studied in association with IBD. Various polymorphisms related to the NOD2 gene have been reported and are known to be related to fibrogenesis in UC and CD [19–23]. The NOD2 gene was also suggested as a predictive marker for the progression of CD fibrosis [24]. Within the innate immune system, Toll-like receptors 4 (TLR) and signal transducers and activators of the transcription 3 (STAT3) might be a mechanism for intestinal fibrosis [19,25,26], and interleukin-23 receptor (IL23R), interleukin-12 subunit beta (IL12B), and Janus kinases 2 (JAK2), which are related to the Th17 pathway, could also be involved [25,27–29]. CX3CR1-mediating chemokines [30] and autophagy genes (autophagy related 16 like 1 [ATG16L1] and immunity-related GTPase family M protein [IRGM]) were reported to have an association with stricture disease [25,31]. However, the exact mechanism remains obscure because fucosyltransferase 2 (FUT2) appears to change the composition of the gut microbiota, which is presumed to be able to induce fibrosis [32]. Transforming growth factor β (TGF-β) plays a broad role in initiating inflammation and fibrosis [33,34]. Matrix metallopeptidase 3 (MMP3) encodes a proteinase that degrades most components of the ECM. Membrane-associated guanylate kinase inverted 1 (MAGI1), which is associated with a mechanism that disrupts the tight junction of intestinal epithelial cells, is a gene factor potentially associated with fibrosis (Table 1) [35,36]. Because the number of studies is too small for generalization, more genome-wide association studies and next generation sequencing studies are needed to reveal genetic factors involved with fibrosis in IBD [16].
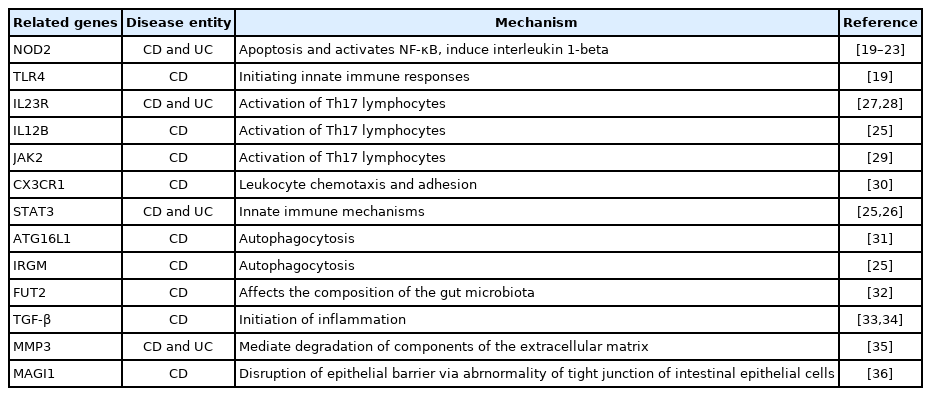
Potential genetic factors of IBD-associated fibrosis
Cytokines and growth factors
Local fibroblasts in fibrotic foci proliferate in response to various growth factors and cytokines. platelet derived growth factor (PDGF), basic-fibroblast growth factor, insulin like growth factor 1, epidermal growth factor, CTGF, tumor necrosis factor α (TNF-α), IL-1β, and IL-6 can act as major proliferating factors [12,14,37]. The proliferating fibroblasts recruit various inflammatory cells, T cells, eosinophils, and mast cells. Inflammatory mediators, such as PDGF-A, PDGF-B, IGF-1, and fibronectin, are also involved in local fibroblastic proliferation and the migration of fibroblasts to the ECM of an inflamed area. In addition, intestinal stellate cells are differentiated into fibroblasts at inflammatory sites using TGF-β [38]. Furthermore, though the molecular pathway is not well established, the capacity of adult bone marrow to derive fibroblast precursors recently became clear, and several cytokines, such as IL-10 or other growth factors, are considered to be part of that pathway [14,39,40].
Critical role of adipose tissue
The role of adipose tissue is essential in inducing hyperplasia of the muscularis propria and subsequent stricture formation in CD [41]. In IBD research, interest is increasing in the role of various bioactive substances secreted by mesenteric fat [42]. Creeping fat is a unique and pathognomonic phenomenon in CD that was first reported by Crohn in 1932 [43]. Creeping fat is defined as > 50% coverage of the exterior intestinal surface with proliferation and ectopic extension of mesenteric adipose tissue (Fig. 1). In the proliferated adipose tissue surrounding the intestine, numerous mediators play crucial roles in inflammation and immunity that lead to the development and progression of IBD [41,44]. Mediators secreted by fat tissue include adipokines (adiponectin, leptin, resistin, C1q/TNF-related protein 3 [CTRP-3], and fatty acids), cytokines (TNF-α, peroxisome proliferator-activated receptor-γ [PPAR-γ], macrophage colony-stimulating factor, monocyte chemoattracted protein-1, IL-1, IL-6, IL-8, IL-10, and chemokine (C-C motif) ligand 5), and growth factors (ghrelin and vascular endothelial growth factor). Those secretions attract and activate various immune cells [45,46]. Therefore, cases of cobblestone mucosa and proper muscle hyperplasia are common in specimens surgically resected to treat CD complications and result in a thick intestinal wall and stricture formation (Fig. 1) [47].
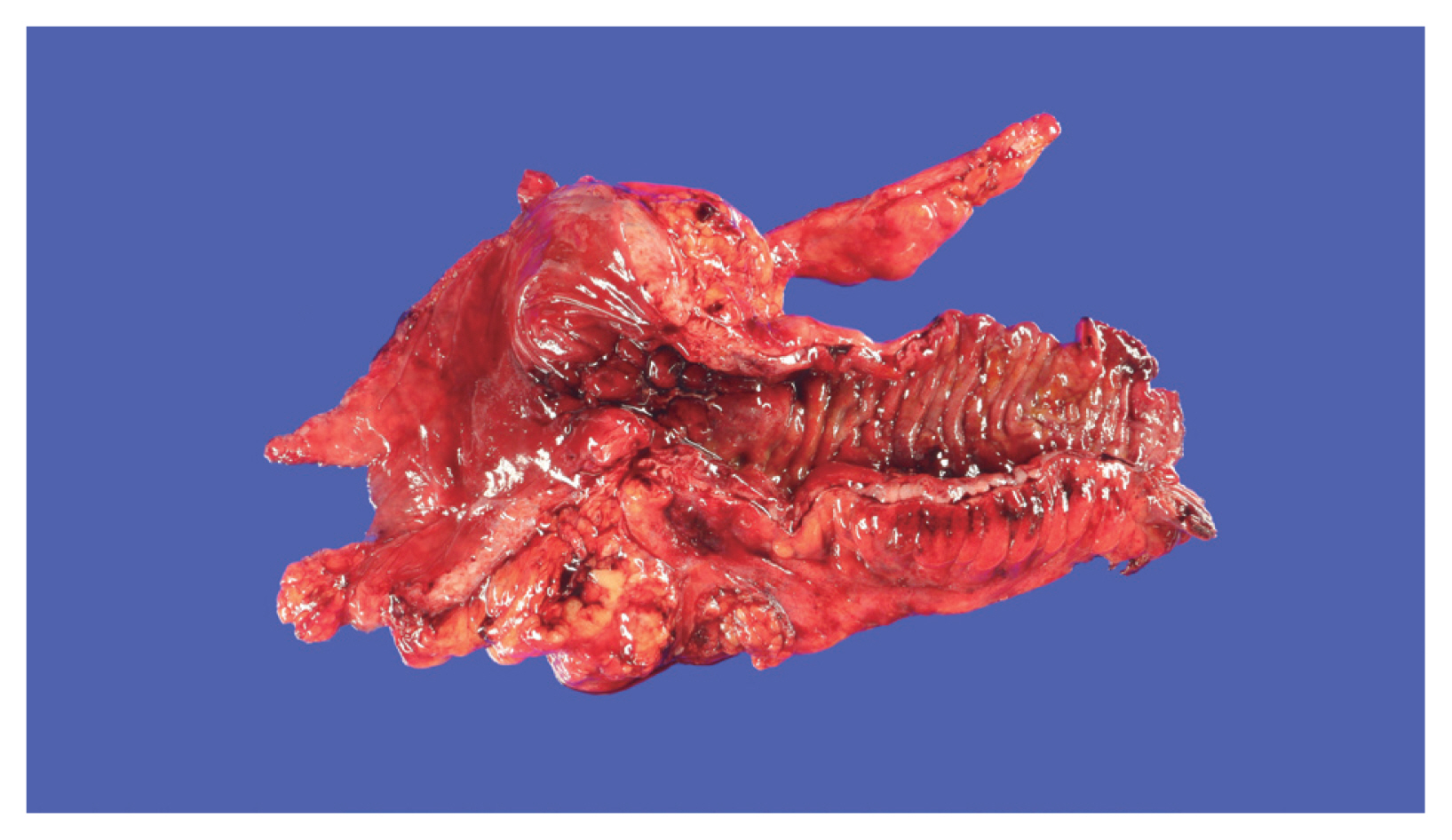
Gross finding of resected large intestine specimen of 30-year-old patient with Crohn’s disease. Creeping fat covers more than 50% of the intestinal circumference surface with mesenteric proliferation. Cobblestone mucosa and proper muscle hyperplasia that result in intestinal stricture are also noted.
EMT and EnMT
The EMT is a well-known phenomenon in malignant neoplasms and literally describes a phenomenon in which tumor cells with epithelial features acquire a mesenchymal tendency to break the resistance of the surrounding ECM, facilitate local migration and invasion, and exhibit aggressive behavior [48]. It is also a mechanism of distant metastasis, in which epithelial cells are attacked by immune cells, or an apoptotic program is initiated when epithelial cells float away from their location, especially when they enter the blood flow, which is the starting point of distant metastases [49].
In IBD, damaged intestinal epithelial cells are activated, and the EMT pathway is initiated. This change can be shown by a loss of epithelial marker expression (such as cytokeratins and E-cadherin) in the enterocytes of inflamed foci and increased expression of mesenchymal markers (especially fibroblast markers, MMP-2, MMP-9, ferroptosis suppressor protein 1, α-smooth muscle actin [α-SMA], and vimentin) [50].
In addition, IBD can damage vascular endothelial cells [14]. In the presence of an excessive inflammatory response, such as hypoxia and secondary mechanical stress, endothelial cells are activated and converted into cells with fibroblast properties. This is called the EnMT, in which endothelial markers (VE-cadherin, Von Willebrand factor, and CD31) expressed in cells are lost, and the expression of fibroblast markers increases. In the EnMT, TGF-β, insulin-like growth factor 2, and IL-1b or TNF-α, which are pro-inflammatory components, are inducers [48,51,52]. Occasionally, this process is reversible. Bone morphogenetic protein-7 or hepatocyte growth factor can convert fibroblasts back into endothelial cells. However, the conditions under which this reversal pathway works remain unknown [53,54]. Cellular transitions, i.e., the EMT and EnMT, are sources of new fibroblasts and result in excessive ECM deposition. Fibroblasts also exhibit enhanced migratory ability. Therefore, the submucosal layer, which is composed of loose connective tissue, is replaced with various ECM components. As a result, the condition of the intestinal wall impedes its flexible movement [13].
Gut microbiota
The gut microbiota is one key factor in the development of fibrosis in IBD [16]. It consists of bacteria, viruses, archaea, protists, and yeast. The composition of the intestinal microbiota affects the host metabolism and immune systems in various ways, and chronic inflammatory status caused by the gut microbiota can ultimately produce the complications of intestinal fibrosis or strictures [55]. The stability of the intestinal microbiota supports the barrier function of intestinal epithelial cells [56]. If the balance between beneficial and harmful bacteria is destroyed, the intestinal microbial barrier and anti-inflammatory regulatory pathway can be damaged, which can eventually cause severe colitis [57,58]. In one study, the intestinal bacterial diversity of mice was reduced using intestinal radiation and an antibiotic cocktail treatment, and that produced decreased levels of TGF, phosphorylated SMAD3, and SMA proteins, which in turn reduced the chronic inflammation that plays a crucial role in intestinal fibrosis [59]. Preliminary studies of fibrosis and the microbiome have been done [24]. Several studies have suggested that fibronectin and collagen deposition in the intestinal wall is a response to bacterial stimulation of the intestine [60,61]. In addition, several studies have reported that strictures are more frequent in patients with CD who have higher levels of antibacterial antibodies [56,58].
HISTOPATHOLOGY
In IBD-associated fibrosis, the change in the muscular layer, which contributes to the presence of a thickened bowel wall, is notable (Fig. 2A). This results in both hypertrophy and hyperplasia [47]. Although chronic inflammation, represented by basal plasmacytosis, is predominant, IBD can also show a mixed inflammation pattern that is accompanied by active inflammation (Fig. 2B, C). As the disease progresses, increased activation of intestinal myofibroblasts results in the gradual synthesis of ECM and contractile proteins (α-SMA and MYLK) [60]. Young fibroblasts begin to be deposited and gradually progress to fibrosis with increased deposition of ECM (collagen, fibronectin, etc.) [55]. In addition, slowed blood flow is caused by damage to highly branched vessels (Fig. 2D). This condition often becomes refractory to medication. Neuronal cell changes are usually observed in surgical specimens from patients with chronic constipation without a certain organic cause, and similarly in IBD patients, neuronal hypertrophy can be a secondary reactive change [62]; however, it can also act as a mechanism for intestinal stiffening (Fig. 2E). In trichrome stain results, dissection between hyperplastic smooth muscle bundles is observed to be interspersed with fibrosis (Fig. 2F).
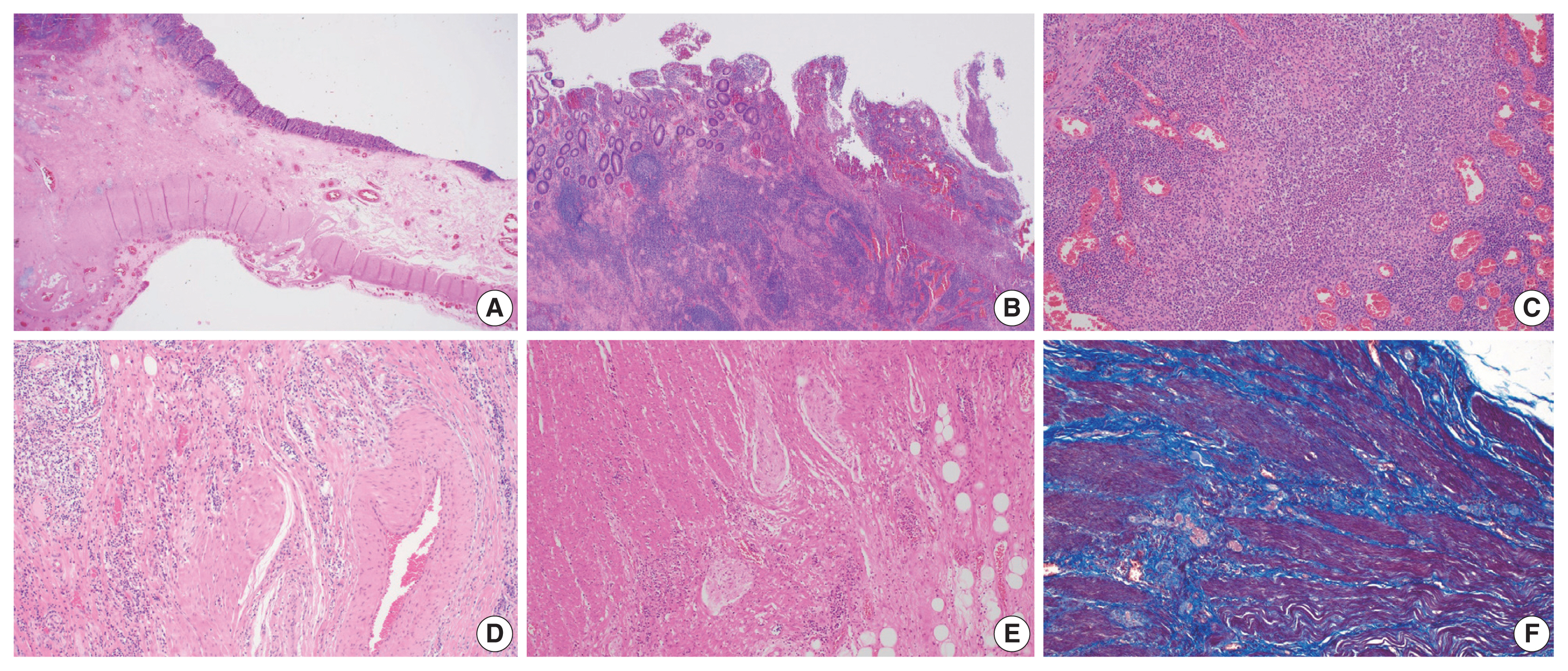
Histologic findings of Crohn’s disease. (A) Hypertrophy and hyperplasia of the submucosa and muscularis propria are present. (B) Chronic inflammation with lymphoid aggregates and lymphoid follicles is dominant. (C) Although chronic inflammation is predominant, active inflammation that consists of extensive neutrophilic infiltrates is also noted. (D) Fibromuscular hyperplasia of damaged submucosal vessels causes slow blood flow. (E) Neural hypertrophy is also noted. (F) Trichrome stain reveals that dissection between the hyperplastic smooth muscle bundles is interspersed with fibrosis.
Histopathology scoring systems for stenosis
In IBD, many scoring systems have been developed to express disease activity, including the Geboes score and Nancy Index of UC and the Crohn’s Disease Activity Index [63,64]. These evaluation methods consider not only the presence of ulcers but also the degree of infiltration of inflammation, submucosal fibrosis, and thickened muscularis propria. Following a recent discussion at the Stenosis Therapy and Antifibrotic Research Consortium, a four-tiered system (none, mild, moderate, and severe) was constructed to include an evaluation of the inflammatory and fibrotic components of each mural layer [55]. In this artificial intelligence era, researchers have endeavored to develop a deep learning model to evaluate intestinal fibrosis in surgical specimens for postoperative recurrence prediction [65,66].
Differences of intestinal fibrosis in UC and CD
UC and CD, which belong to the same IBD category but differ in their mechanisms of development and clinical features, also show differences in IBD-related fibrosis [67]. In UC, fibrotic changes are limited to the mucosal and submucosal layers [55]. This can shorten or stiffen the intestine, leading to motility disorders. Because strictures in UC are rare and can be either benign or malignant, a persistent stricture should raise suspicions of cancer [68]. Complications that are mainly due to bowel wall thickening, such as stricture and stenosis, are problematic in CD. Furthermore, diffuse transmural collagen layers down to the muscularis propria and proliferative fibroblastic infiltration are observed. In UC, on the other hand, the progression of intestinal fibrosis does not correlate with disease duration or location; however, inflammatory activity does correlate with medical treatment. In contrast, in CD, the duration and location of the disease and type of treatment are related to the risk of intestinal fibrosis [55,69,70].
Biomarkers and potential antifibrotic agents
Gene variants, epigenetic modifiers, antimicrobial antibodies, ECM components, and clinical, endoscopic, or environmental factors can be used to evaluate and predict fibrosis in IBD patients [71]. Fibrosis in CD, which causes several serious sequelae, is reversible, and thus it is important to develop therapeutic agents targeting it [72]. However, no effective therapeutic agents are available to prevent or repair the progression of fibrosis except by suppressing inflammation, though diverse potential antifibrotic therapies have been proposed. Although their mechanism is still unknown, statins (simvastatin) have been reported to effectively inhibit the progression of CD fibrosis [73,74]. In CD, pirfenidone, Rho kinase inhibitors, TGF-β signaling inhibitors, IL-13 inhibitors, and G31P (an antagonist of CXCL8) are also known to be effective [75,76]. GED-0507-34, an agent with a strong affinity for PPAR-γ, has been suggested as an anti-fibrotic agent in UC patients [77].
CONCLUSION
IBD-related intestinal fibrosis is the starting point for serious complications in patients with refractory and poorly controlled chronic IBD. If the uniquely activated profibrotic pathway observed in IBD can be identified, it could be used as a biomarker for targeted therapy. In addition, the gene expression signature of fibrogenesis at diagnosis could predict the risk of surgery. Intestinal fibrosis is an unfavorable result of the harmonic action of intestinal epithelial cells, the microbiota, and various mesenchymal components, such as the adipose tissue, fibroblasts, smooth muscle, neural tissue, and vascular endothelial cells, at the lesion site. It is thus necessary to pay attention to these mechanisms, from analyzing the ECM to developing therapeutic agents that target the main factors affecting pathogenesis, as well as elucidating the mechanisms involved by using various advanced research methods.
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
All data generated or analyzed during the study are included in this published article (and its supplementary information files).
Code Availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Author Contributions
Conceptualization: SUB. Figures and data: JMP. Writing—original draft: JMP, JK, YJL. Writing—review & editing: HWL. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.