Radiotherapy Response in Microsatellite Instability Related Rectal Cancer
Article information
Abstract
Preoperative radiotherapy may improve the resectability and subsequent local control of rectal cancers. However, the extent of radiation induced regression in these tumours varies widely between individuals. To date no reliable predictive marker of radiation sensitivity in rectal cancer has been identified. At the cellular level, radiation injury initiates a complex molecular network of DNA damage response (DDR) pathways that leads to cell cycle arrest, attempts at re-constituting the damaged DNA and should this fail, then apoptosis. This review presents the details which suggest the roles of DNA mismatch repair proteins, the lack of which define a distinct subset of colorectal cancers with microsatellite instability (MSI), in the DDR pathways. Hence routine assessment of the MSI status in rectal cancers may potentially serve as a predictor of radiotherapy response, thereby improving patient stratification in the administration of this otherwise toxic treatment.
Preoperative radiotherapy for rectal cancer
Akin to other solid tumours, complete surgical resection remains the mainstay of curative treatment in colorectal cancers (CRCs). This is comparatively simpler in localized colonic lesions, surrounded by an enveloping free serosal surface. However, approximately a third of CRCs arise in the rectum,1,2 where close proximity circumferentially to other pelvic organs and the sacrum, and for low lying examples, inferiorly to the anal sphincter can either limit operative access or require extensive surgical resection. This is attested by poor improvements in the overall survival of rectal cancer sufferers, relative to those with colonic tumours.1
To improve the probability of complete surgical resection of rectal cancer with preservation of the anal sphincter, pre-operative radiotherapy with or without chemotherapeutic agents may be utilized to reduce the tumour volume. Even in the context of modern total mesorectal excision surgery,3,4 pre-operative radiotherapy has shown to provide an additional benefit in the local control of rectal cancer, more effective than post-operative radiotherapy.5-7
Unpredictability of radiotherapy response
Based on the above and other8,9 studies, it is less conclusive whether pre-operative radiotherapy also improves overall patient survival. The latter may reflect the lack of influence of pre-operative radiotherapy on the progression of metastatic disease9 and/or the wide variations in the treatment effect seen in practice between individual tumours.
The inconstant extent of tumour regression in irradiated rectal cancers, even amongst microscopically and clinically identical examples following the same radiation schedule, has been the subject of various grading systems.10-15 Although these differ in their nomenclature (for example, tumour regression grade12,15 vs residual tumour cell density10), number of tiers and their cut-offs, they all aim to microscopically quantify regression as areas of fibrosis within tumour that are devoid of cancer cells. As well as the lack of standardization, issues regarding the reproducibility of regression grading have been previously highlighted.16 Putative contributing factors include inconsistency in the volume of tumour sampled for grading and the difficulty in distinguishing between tumour induced stromal desmoplasia, especially at the leading invasive tumour edge, with radiotherapy induced fibrosis replacing eradicated cancer cells.
Despite these reservations, several rectal cancer cohorts treated with pre-operative radiotherapy have shown a relationship between histological grading of tumour regression and survival outcome.17-20 Accordingly, the latest version of the Cancer Staging Manual from the American Joint Committee on Cancer recommends that while evidence is not conclusive, the extent of tumour regression should nonetheless be graded in radiotherapied rectal cancers.21
Predictive markers of sensitivity to radiotherapy
The capricious nature of radiotherapy induced rectal cancer regression is an issue, with possible short term side effects of radiation ranging from nausea, diarrhea and skin erythema to acute urinary retention, proctitis, thromboembolic disease and lumbosacral plexopathy.22 Patients who received pre-operative radiotherapy for rectal cancer have also reported significantly increased rates of chronic fecal incontinence23 and sexual dysfunction.24 The capacity of these toxic side effects to considerably diminish the patient's quality of life necessitates a reliable means of predicting the likely benefit of radiotherapy in individual cases.
But finding such marker(s) has proved difficult. Candidate molecules with a potential to fulfil this role include those involved in the cellular DNA damage response (DDR) pathways, such as apoptosis related bcl-225 and survivin,26 as well as DNA double strand break (DSB) recognizing Ku80,27 and those that potentiate the malignant tumour phenotype, such as epidermal growth factor receptor (EGFR)28 and vascular endothelial growth factor.25 However, none of these or others has as yet convincingly shown the clinical applicability as a predictive marker of radiotherapy response. Analyses using oligonucleotide microarrays have also provided inconsistent gene profiles of radiation sensitivity in rectal cancer.29-31
Microsatellite instability (MSI) status as predictor of sensitivity to radiotherapy
Only a few clinical studies have examined the MSI status in CRC as a marker of radiotherapy response.32 Although this relationship could not be demonstrated, these studies involved small cohorts (n<100) where the number of MSI related CRC cases are expected to be low.33 There is however accumulating molecular evidence of the various roles that DNA mismatch repair (MMR) proteins, the lack of which define the MSI phenotype, contribute to the molecular processes in exposed cells which attempt to repair and survive radiation injury. Before presenting these details, both the cellular response to radiotherapy and the genetic, clinical and pathological aspects of MSI related CRCs will be outlined.
CELLULAR RESPONSE TO RADIOTHERAPY
Radiotherapy delivers ionizing radiation to target cells, aiming to induce their death by inflicting a variety of damage to their genome. Amongst these, DSBs of the chromosomal DNA are regarded as the most lethal form of injury, where both strands of the DNA helix are severed. In response, a highly complex and incompletely understood network of intracellular molecular pathways is triggered, collectively described as the DDR.
General scheme of DDR
DDR can be triggered by both exogenous and endogenous factors, including radiotherapy, reactive oxygen species from normal cell metabolism and replication errors during cell division.34 Subsequently activated molecules can be broadly grouped into the sensors, transducers and effectors of the DDR.35 The sensor proteins of DNA damage include the meiotic recombination 11 (MRE11) complex, a trimer of MRE11, RAD50, and NBS1 (also known as XRS2) molecules.36 The role of DDR transducers in humans are centrally played by the two phosphatidylinositol 3-kinase-like proteins ataxia telangiectasia mutated (ATM, also known as TEL1) and ataxia telangiectasia and RAD-3 related.37,38 These proteins relay the damage signal to a variety of DDR effectors by controlling their phosphorylation, which result in the simultaneous arrest of the cell cycle and attempted repair of the DNA damage, proceeding to apoptosis should this fail. Examples of such effector proteins include checkpoint kinase 1 and 2 (CHK1 and CHK2, respectively) that leads to cell cycle arrest and p53 influencing both cell cycle arrest and apoptosis.34 Hence those cancer cells that are deficient in the functional constituent protein(s) of this response may be especially prone to death from DNA damage inducing agents, such as radiation.38
DDR specific to radiation injury
The potential for radiation induced DNA DSBs alone to cause cell cycle arrest is low. The signal is instead enhanced by the binding of MRE11 complex to the DSBs, with generation of regions of single stranded DNA by its nuclease activity. This in turn recruits and stimulates ATM (TEL1) that amplifies the checkpoint signalling and cell cycle arrest.36
Of the different repair pathways that are tailored to specific types of DNA injury, the two important mechanisms in the context of DNA DSBs are homologous recombination and non-homologous end joining (NHEJ). The former pathway requires a homologous sequence of DNA from the sister chromatid or another chromosome to serve as either a guide or a donor to bridge the breaks. By contrast, NHEJ ligates the broken ends of the DSBs directly without the need of a homologous DNA strand template. NHEJ represents the main mechanism by which non-cell replication associated DSBs are repaired, as in those induced by ionizing radiation.38
In NHEJ, DSB is initially recognized by binding of the Ku70/80 heterodimeric protein to the exposed DNA ends. DNA-dependent protein kinase catalytic subunit (DNA PKcs) is then attracted to the DSB, which activates its kinase activity. This leads to the recruitment of other molecules, such as DNA ligase IV, that process and rejoin the DNA ends.38,39 There is also evidence that the MRE11 complex signalling the cell cycle arrest may also be involved in the NHEJ following DNA DSBs.40
CRC WITH MSI
Molecular basis of MSI
Microsatellites are tandemly repeated short DNA motifs throughout the genome. Apart from stable heritable inter-individual polymorphisms, their lengths are conserved. But microsatellite lengths can be altered by single strand damage that leads to DNA mismatch replication errors involving nucleotide base insertions and/or deletions during cell division. Thus initiated DNA MMR pathway recognizes the error and co-ordinates the substitution with correct base pairing. In eukaryotes, the key proteins involved in this process are MLH-1, MSH (such as MSH-2 and MSH-6), and PMS-2.41
The failure to restore these errors in cell clones (i.e., tumours) with defective MMR results in discordant lengths of the corresponding microsatellite loci between the tumour cell genome and that of the normal cells of the same individual.42 Such MSI accounts for approximately 15% of all CRCs.
Chromosomal instability (CI) and MSI represent two recognized models of genomic instability in the genesis of CRC. In the majority of cases, tumour cells demonstrate CI with aneuploidy, with structural and/or numerical chromosomal abnormalities. Based on Vogelstein et al.'s43 classic colorectal adenoma-carcinoma sequence, this 'gatekeeper pathway' involves the loss or mutation of several key genes including adenomatous polyposis coli affecting nuclear localization of the transcription factor β-catenin, KRAS integral to the EGFR pathway, SMAD4 signalling transforming growth factor β (TGFβ) mediated apoptosis, CDC4 regulating the G1/S transition of the cell cycle and p53, one of the key mediators of apoptosis and cell cycle arrest.44
The remaining CRCs instead harbour MSI with tumour cells exhibiting diploid or near diploid chromosomes.45 In this 'caretaker pathway', genes implicated in the 'gatekeeper pathway' are infrequently altered and traditional adenomas are uncommonly seen. Instead, an alternative, analogous sequence of molecular alterations is hypothesized, including mutations of WNT in β-catenin signalling, BRAF lying downstream to KRAS, TGFβ receptor 2, pro-apoptotic BAX, and insulin like growth factor-2 receptor.44
Clinical and pathological features of CRC with MSI
MSI CRCs are usually sporadic, with methylation of the promoter region of MLH-1. In a subset of cases, germline mutation of MLH-1 and/or MSH-2, MSH-6 or rarely PMS-2 is seen as hereditary non-polyposis colorectal cancer (HNPCC; Lynch syndrome).42 The latter manifests clinically with autosomal dominant expression of both colorectal and other cancers, including those of primary endometrial, gastric and ovarian origin, at an early age of onset before the mid to late 40's.45
MSI CRCs bear certain features with increased frequency. These include proximal location, poorly differentiated or undifferentiated tumours, tumour infiltrating lymphocytes, Crohn's disease-like host response with lymphoid aggregates and mucinous, signet ring or medullary type histology.45,46 Furthermore, the evidence is strong that CRCs with MSI have a better prognosis, as highlighted in a large meta-analysis with hazards ratio for death of 0.65 in comparison to microsatellite stable cases.47
Susceptibility to chemotherapy may also be altered in CRCs with MSI. Experimental data supports the theory that a competent DNA MMR pathway is required to facilitate the actions of certain therapeutic agents. For instance, the pyrimidine analog 5-fluorouracil (5-FU) acts by incorporating into RNA and DNA, as well as by inhibiting thymidylate synthase required for thymidine nucleotide production.48 Its cytotoxicity appears to depend on the recognition and processing of the incorporated 5-FU by the MMR process.49 Clinical trials appear to concur with this, showing that the survival of MSI CRC patients with low stage disease do not substantially improve with adjuvant 5-FU therapy, though the benefits of same treatment to non-MSI related low stage CRC cases appear to be small.50,51
Assessment of MSI status
The MSI status of CRCs can be assessed by either immunohistochemistry (IHC) to highlight the expression of DNA MMR proteins in tumour cells, or by polymerase chain reaction (PCR) amplification and comparison of the lengths of the microsatellite loci in matching tumour and normal cell chromosomes. The two methods are comparable in their ability to identify cases with MMR protein mutations,52 though the sensitivity of PCR based testing appears to be slighter higher.53 IHC is however simpler, better suited to allow for pre-operative consideration of the MSI status from colonoscopic tumour biopsies in a clinically appropriate timeframe. Decisions regarding the extent of bowel resection and the possibility of concurrent hysterectomy and oophorectomy can thus be made expeditiously, especially in patients with raised clinical suspicion of HNPCC.54 In such cases, IHC has the additional advantage of directing the subsequent mutation analysis to a specific protein. PCR may follow IHC if the latter contradicts the clinical impression and suggests a functional DNA MMR. In patients with low clinical suspicion of HNPCC, either method may be adequate as the first line of testing. If the result is consistent with MSI, then mutation analysis of the MMR protein(s) should follow.53
Early reports (e.g., Thibodeau et al.55) and majority of large studies since then (e.g., Lindor et al.54) have used a 2 panel antibody for IHC testing, targeting MLH-1 and MSH-2. Others have however included MSH-6 in this panel, as its mutations affect an important subset of HNPCC patients and as MSH-2 functions as a heterodimer with MSH-6.56 Similarly, MLH-1 acts as a heterodimer with PMS-2 in DNA MMR, and the loss of MLH-1 abrogates this protein complex. Thus the expression status of PMS-2 should correspond to that of MLH-1, and the inclusion of PMS-2 in the IHC panel should increase the sensitivity of MSI detection by identifying those additional cases with MLH-1 mutations which, for a number of reasons, may show false positive staining for MLH-1 but are negative for PMS-2.57 However, the concept of a substantial role of PMS-2 in the pathogenesis and detection of HNPCC and sporadic MSI tumours appears to be still evolving.58
The 2004 revised Bethesda guidelines for the selective MSI testing of CRC patients is based on the above clinical and pathological features. Positive family history of any MSI/HNPCC related cancers at young age is included in the criteria to account for germline mutation related cases. But these features have been less than reliable in the identification of CRCs with MSI.45 Others instead advocate for the routine testing of DNA MMR status in all CRCs as a prognostic indicator and possibly predictive marker of chemotherapy response.59
POSSIBLE LINKS BETWEEN MSI AND RADIATION SENSITIVITY IN CRC
There is accumulating evidence to suggest that DNA MMR proteins may influence and/or are directly involved in the DDR following radiation induced DSBs. That is, their deficiency that characterizes MSI CRC cancers may also indicate sensitivity to radiotherapy.
The hypothetical link is plausible at least in that an underlying DNA MMR defect will contribute to a greater instability of the genome, allowing for the aggregation of genetic mutations involving the necessary components of the relevant DDR pathways. For instance, reports have demonstrated increased rates of mutations in MSI tumour cells involving proteins such as ATM60 and MRE1160,61 that are integral to the recognition and downstream signalling following DNA DSBs, and DNA PKcs62 required for the repair of the DSB ends by NHEJ.
MSI cancers may exhibit radiation sensitivity also because of possible direct role(s) that MMR proteins play in the DDR to ionizing radiation, implicated at multiple stages in the pathways affecting cell cycle arrest, subsequent DSB repair and apoptosis.63 This could include the participation of MLH-1 and MSH-2 in the initial recognition of radiation induced DNA damage.64,65 Moreover, MSH-2 negative cells fail to mobilize MRE11 and show increased death following radiation.66 Both MLH-1 and MSH-2 additionally appear to facilitate efficient arrest of the cell cycle at the G2/M phase transition during the DDR.64,66 As well, experimental evidence in human fibroblasts suggest that MLH-1 and PMS-2 are molecular targets for p53, the effector of both cell cycle arrest and apoptosis in DDR, and may guide the direction of p53 mediated signalling between these two processes.65
Loss of MLH-1 also correlates with the lack of activation of nuclear factor-κB following DSBs,67 an effector in the DDR cascade downstream to ATM after ionizing radiation exposure and which promotes cell survival by counteracting the p53 mediated apoptotic signal.68 These MMR proteins may furthermore contribute to the accuracy of the DNA repair process, by modulating the correct nucleotide base pairing at the DSB ends as they are bridged by the error prone NHEJ pathway.69
Thus as summarized in Fig. 1, MLH-1 and MSH-2 may be directly or indirectly involved in multiple steps of the DDR following radiation injury, beyond the role of DNA MMR. This may encompass the whole spectrum of DSB related DDR, from detection of the DSBs to the subsequent mediation of signalling by the transducer molecules, induction and maintenance of the cell cycle arrest, actual repair of the DSBs and the interim inhibition of apoptosis, assurance of the integrity of the repaired DNA prior to resumption of the cell cycle and the switch to cell death should the DSB repair fail.
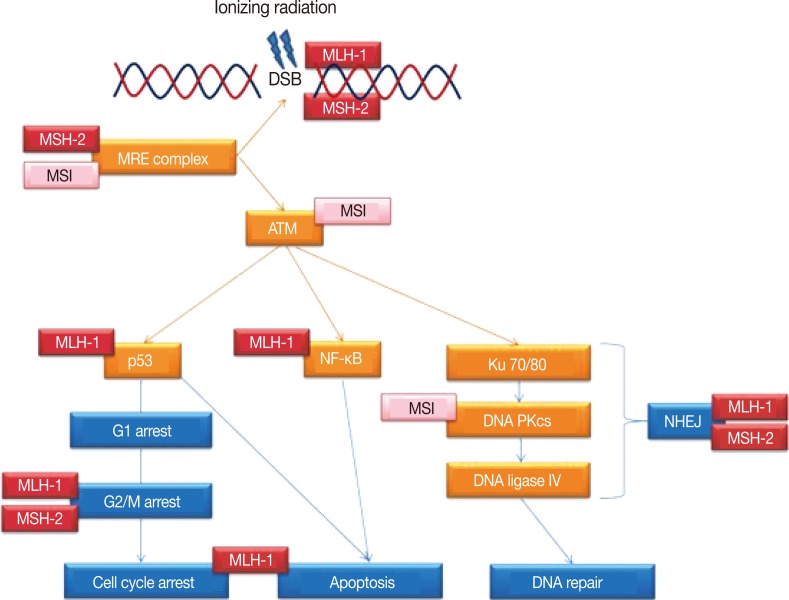
Simplified schematic representation of the DNA damage response pathways following ionizing radiation exposure leading to DNA double strand breaks (DSBs). Indicated are the multiple steps/proteins at which either microsatellite instability (MSI) of the genome or DNA mismatch repair proteins MLH-1 and MSH-2 may affect this response. ATM, ataxia telangiectasia mutated; NF-κB, nuclear factor-κB; DNA PKcs, DNA-dependent protein kinase catalytic subunit; NHEJ, non-homologous end joining.
CONCLUSION
Pre-operative radiotherapy improves local disease control and possibly overall survival in rectal cancer sufferers. These benefits however vary considerably between individuals. Means of predicting a positive response will allow for the unnecessary administration of this potentially toxic treatment to be avoided. But such predictive markers are currently unavailable.
Sequencing of the human genome and identification of approximately 22,000 protein encoding genes,70,71 combined with technological advances in microarray profiling has led to a proliferation of data or 'omics' driven research. Some of these have applied to radiation sensitivity of rectal cancer, producing incompatible results.29-31 This is perhaps because such projects are conducted primarily to generate a large amount of data in which specific patterns are then sought, rather than testing a preconceived hypothesis. While attracting some criticism for this reason,72 data driven studies may still provide extensive results that inspire new specific directions in experimental designs to a select number of gene(s).73
But in the form of DNA MMR proteins, we already have a robust set of markers that are routinely assessed in CRCs in clinical practice, with established roles in the identification of MSI/HNPCC related cases and in patient prognostication. Their expression status is easily assessed with IHC. Available molecular evidence suggests multiple roles of these proteins beyond MMR, specifically within the pathways of DDR following ionizing radiation induced DSBs. This hypothesis can be tested in a comparably simple manner, in a large number of pathological samples of paraffin embedded rectal cancer tissues with IHC and correlated with both histological tumour regression in the resected bowel following radiotherapy and with survival outcomes. Further research should aim to translate the presented experimental data to clinically obtained rectal cancer tissues, and if proven right, the assessment of MSI status in tumour biopsies from colonoscopy may hold the answer to not only the patient prognosis, but also their sensitivity to radiotherapy.
Notes
No potential conflict of interest relevant to this article was reported.