Rosai-Dorfman Disease: Report of a Case Associated with IgG4-Related Sclerotic Lesions
Article information
Abstract
We describe a rare case of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) associated with a six-year history of autoimmune pancreatitis, which was controlled by steroid treatment. The patient presented with multiple, cervical and thoracic lymphadenopathy and abnormal, nodular opacities in the lung. Histologically, Rosai-Dorfman disease with numerous IgG4-positive cells was identified in a subcutaneous lymph node in the patient's left forearm. The patient recovered uneventfully with steroid treatment.
Rosai-Dorfman disease (RDD), a sinus histiocytosis with massive lymphadenopathy, represents an idiopathic proliferation of peculiar, histiocyte-like cells of unknown etiology. Most patients with RDD present with multiple lymphadenopathy, although RDD also involves a variety of extranodal sites. IgG4-related sclerotic disease is a recently described entity, showing sclerosis with the presence of IgG4-positive plasma cells. It is currently thought that RDD may belong to the disease spectrum of IgG4 sclerotic disease. Here we report a case of RDD in a 70-year-old Korean male with a history of autoimmune pancreatitis.
CASE REPORT
A 70-year-old male patient was diagnosed with autoimmune pancreatitis in May 2004. At that time, the serum IgG was mildly increased to 1,780 mg/dL (normal, 694 to 1,618 mg/dL). Until October 2009, he had been receiving steroid treatment, and he had a history of recurrence of autoimmune pancreatitis in November 2005.
In July 2010, a follow-up chest computed tomography was performed and showed abnormal findings with multiple enlarged lymph nodes in the bilateral mediastinal and both hilar areas as well as a small irregular nodular and patchy ground-glass opacity in both of the upper lobes and in the right lower lobe of the lung (Fig. 1). A physical examination showed a 2 cm skin nodule on his left forearm. The laboratory findings were as follows: IgG, 1,900 mg/dL (normal, 694 to 1,618 mg/dL); IgG4, 3.56 g/L (normal, 0.06 to 1.214 g/L); and serum amylase, 258 U/L (normal, 30 to 110 U/L).
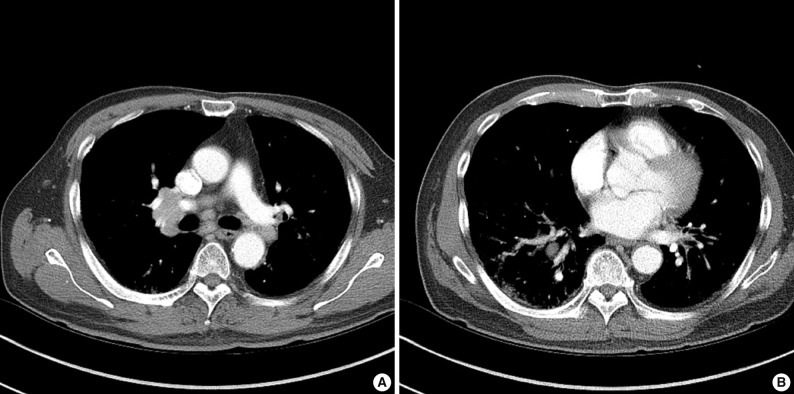
(A) Chest computed tomography (CT) shows multiple, lymph node enlargement in the bilateral mediastinal and hilar areas. (B) Chest CT shows multiple irregular nodular opacities and peribronchovascular infiltration in both lungs.
A biopsy of the skin nodule revealed that it was a subcutaneous lymph node (Fig. 2). This lymph node showed effacement of the lymph node structure with a dense lymphoid infiltrate with marked sinus expansion by histiocyte-like cells with intermediate to large round nuclei, vesicular chromatin, and ample cytoplasm containing small phagocytosed lymphocytes, red cells, and plasma cells (emperipolesis). The plasma cells were abundant both in the sinus and in the medullary cords. The immunohistochemical stain showed that the histiocytes were positive for CD68 and S-100 protein. Numerous IgG4-positive cells (approximately 200 IgG4 cells/high power field) were also noted. A bronchoscopic biopsy of the peribronchial lymph nodes was performed and showed a few non-caseating granulomas that were suspicious for dust granulomas with approximately 20 IgG4-positive plasma cells. The patient recovered with steroid therapy.
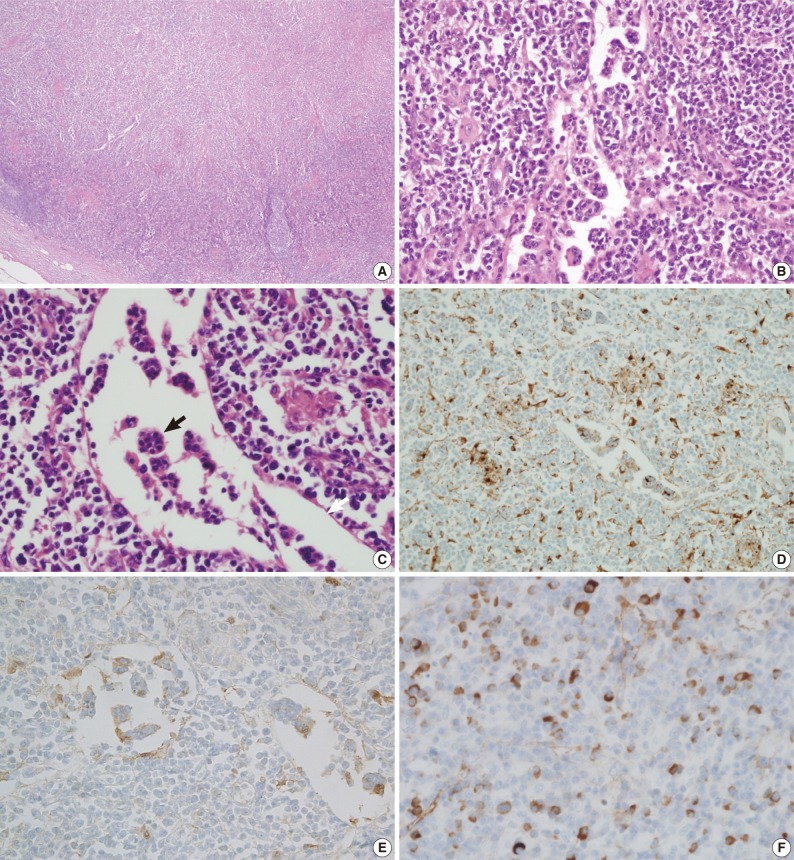
Microscopic findings of a subcutaneous lymph node in the left forearm. The lymph node structure is effaced (A) and composed of sheets of histiocytoid cells, plasma cells, and lymphocytes (B, C) with emperipolesis (black arrow) in the dilated sinuses (white arrow). Immunohistochemical staining shows that the histiocytoid cells are immunopositive for CD68 (D) and S-100 protein (E). A significant proportion of the plasma cells are positive for IgG4 (F).
DISCUSSION
RDD is a rare disease of benign histiocytic proliferation of unknown etiology. It usually affects lymph nodes, but can also involve a variety of extranodal sites including the skin, lungs, breast, and bone.1-5 Histologically, the lymph node architecture is altered by massive dilatation of the sinuses and effacement of follicles with histiocyte-like cells showing findings of lymphophagocytosis and emperipolesis.1 Clinically, RDD in lymph nodes has been known to be self-limited, although extranodal or multifocal RDD has various outcomes.3 Although the etiology of RDD is still unknown, it was recently reported that RDD may be associated with IgG4 sclerotic disease.2-4,6
IgG4-related sclerotic disease is a recently described entity with multi-organ involvement including the pancreas, salivary gland, orbit, hepatobiliary tract, retroperitoneum, thyroid, mediastinum, pleura, mucosa of gastrointestinal tract, and lymph nodes. The disease is characterized by increased serum IgG and IgG4 levels and the presence of many IgG4-positive plasma cells. Clinicians are commonly suspicious for a malignant tumor because it usually presents in the form of mass lesions. The pathological findings have shown lymphoplasmacytic infiltration, sclerosis, and/or obliterative phlebitis. Clinically, the disease is characterized by a good response to steroid therapy.7-9 In the present case, the patient had a history of autoimmune pancreatitis. The pancreas is known to be the most commonly involved organ in IgG4-related sclerotic disease. Autoimmune pancreatitis (lymphoplasmacytic sclerosing pancreatitis) is a distinct type of chronic pancreatitis, and it is now widely accepted as one of the IgG4-related sclerotic disease.8
Several reports have studied the possible relationship between RDD and IgG4-related sclerotic disease. Kuo et al.4 reviewed 12 cases of cutaneous RDD (CRDD) and found that CRDD showed histopathological features similar to those of IgG4-related sclerotic disease. Many IgG4-positive plasma cells are observed in CRDD cases, which suggest that CRDD may be related to IgG4-related sclerotic disease.4 Roberts and Attanoos2 also reported a case of pulmonary RDD with a significantly increased number of IgG4-positive plasma cells and suggested a possible overlap between RDD and IgG4-related sclerotic disease.2 Shrestha et al.6 studied the histopathology of pulmonary IgG4-related disease with increased IgG4-positive plasma cells and autoimmune pancreatitis. They observed characteristic pulmonary findings in the lung of autoimmune pancreatitis including endothelialitis, fibrosis, lymphangitic inflammatory infiltrates with plasma cells, and histiocytes. They also examined other various diseases, including Erdheim-Chester disease, pulmonary Sjögren syndrome, inflammatory myofibroblastic tumor, and nodal or extranodal RDD, to compare to the histologic findings of pulmonary IgG4 disease. Among these other diseases, 8 of the cases were nodal or extranodal RDD. Six out of the 8 cases showed increased IgG4-positive cells and a ratio of IgG4/IgG (range, 10 to 70%). Two out of the 3 nodal RDD cases had an IgG4/IgG ratio of 40% and 70%. The authors suggested further study to determine the possible correlation with IgG4-related sclerotic disease and RDD.6 Richter et al.3 described a case of extranodal cardiac RDD, and also investigated its possible association with IgG4-related sclerotic disease. However, the number of IgG4-positive plasma cells within the infiltrates was not increased in that case, therefore, they did not conclude that this type of RDD belongs to the disease spectrum of IgG4-related sclerotic disease. These three previous reports have not shown any evidence of other organ involvement with IgG4-related sclerotic disease in comparison to the present case.
In conclusion, we observed a unique case of RDD with marked increase of IgG4-positive plasma cells in a patient with a history of autoimmune pancreatitis. This may suggest a possible close relationship of RDD and IgG4 sclerosing disease, although further investigation with a larger number of cases will be necessary for determining the relationship between RDD and IgG4-related sclerotic disease.
Notes
No potential conflict of interest relevant to this article was reported.