E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 58(2); 2024 > Article
-
Case Study
Malignant potential of neuroendocrine microtumor of the pancreas harboring high-grade transformation: lesson learned from a patient with von Hippel-Lindau syndrome -
Jongwon Lee1
 , Kyung Jin Lee2, Dae Wook Hwang3, Seung-Mo Hong,1
, Kyung Jin Lee2, Dae Wook Hwang3, Seung-Mo Hong,1 -
Journal of Pathology and Translational Medicine 2024;58(2):91-97.
DOI: https://doi.org/10.4132/jptm.2024.02.13
Published online: March 13, 2024
1Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
2Department of Radiology and Research Institute of Radiology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
3Department of Surgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding Author: Seung-Mo Hong, MD, PhD, Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-4558, Fax: +82-2-472-7898, E-mail: 'smhong28@gmail.com'
© 2024The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 656 Views
- 118 Download
Abstract
- Pancreatic neuroendocrine microtumor (PNEMT) is a neuroendocrine tumor (NET) < 0.5 cm in diameter, and it is considered benign. We report a PNEMT with high-grade transformation (HGT). A man in his 60s with von Hippel-Lindau syndrome underwent surgical resection of a NET. A second sub-centimeter nodule with a nodule-in-nodule pattern was discovered. The 0.4 cm outer nodule contained clear columnar cells with round nuclei and indistinct nucleoli, while the 0.1 cm inner nodule had eosinophilic cells with an increased nuclear to cytoplasmic ratio, vesicular nuclei, and prominent nucleoli. Tumor cells in the outer and inner nodules were synaptophysin and chromogranin positive. Only the inner nodule was p53 positive, while the outer nodule was exclusively positive for carbonic anhydrase 9 and vimentin. The Ki-67 labeling indices for the outer and inner nodules were 2.1% (grade 1) and 44.3% (grade 3), respectively. This nodule was determined to be a PNEMT with HGT. Our findings suggest that a PNEMT may not always be benign and can undergo HGT.
- Clinical features
- A male patient in his 60s with clinically and genetically confirmed VHL syndrome visited for a biannual checkup. He had undergone previous surgeries, including a pylorus-preserving pancreaticoduodenectomy (PPPD) for grades 1 and 2 PanNETs and a macrocystic serous cystic neoplasm; a left partial nephrectomy for a clear cell type renal cell carcinoma and renal cysts; bilateral subtotal adrenalectomies for bilateral adrenal pheochromocytomas and a right adrenal cortical adenoma.
- The patient’s height and weight were within normal ranges. At the time of admission, all of his vital signs, including blood pressure, heart rate, respiratory rate, and body temperature, were within the normal range. His electrolyte levels were also within normal ranges. There were no noticeable signs of hormonal overexpression.
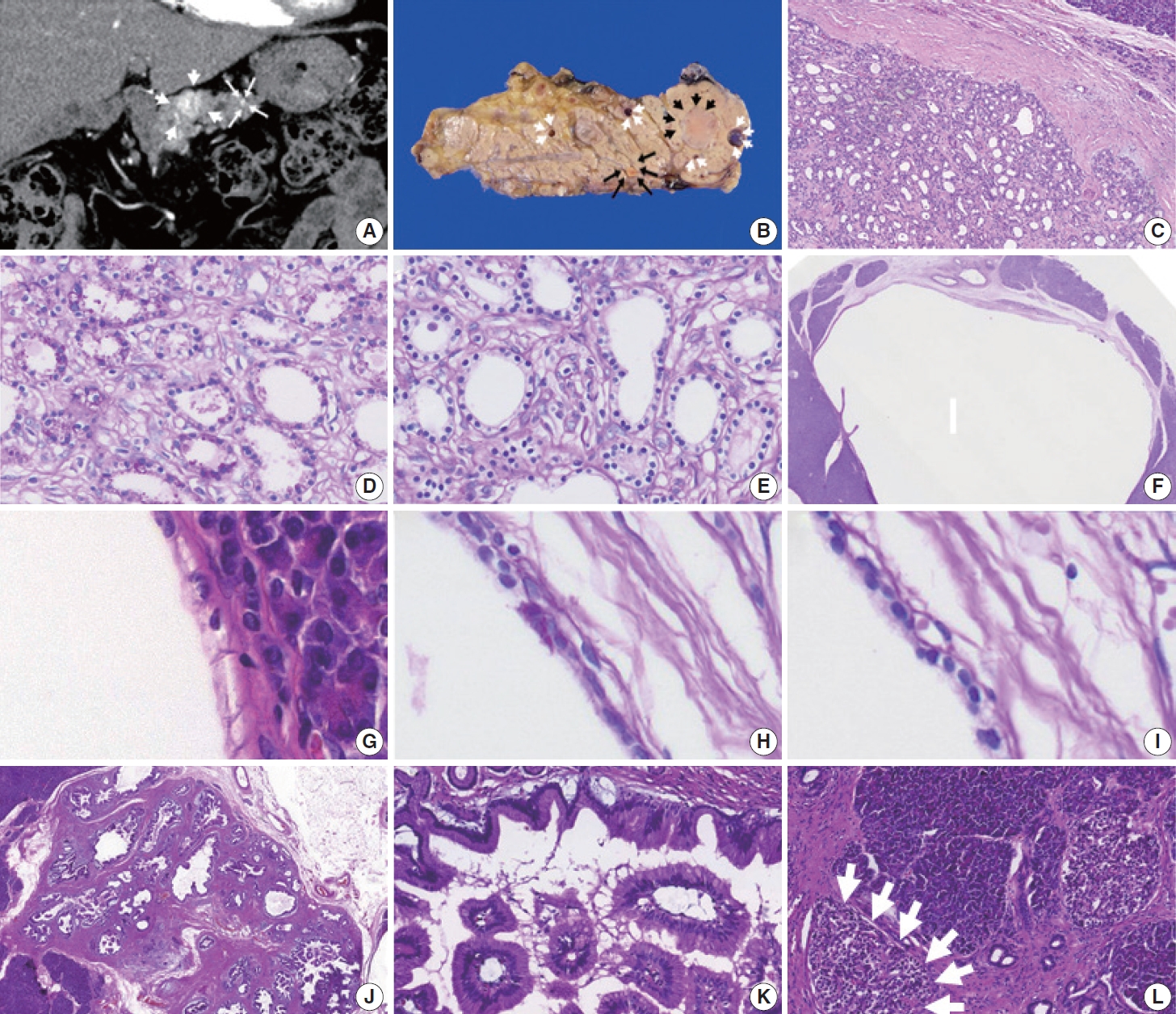
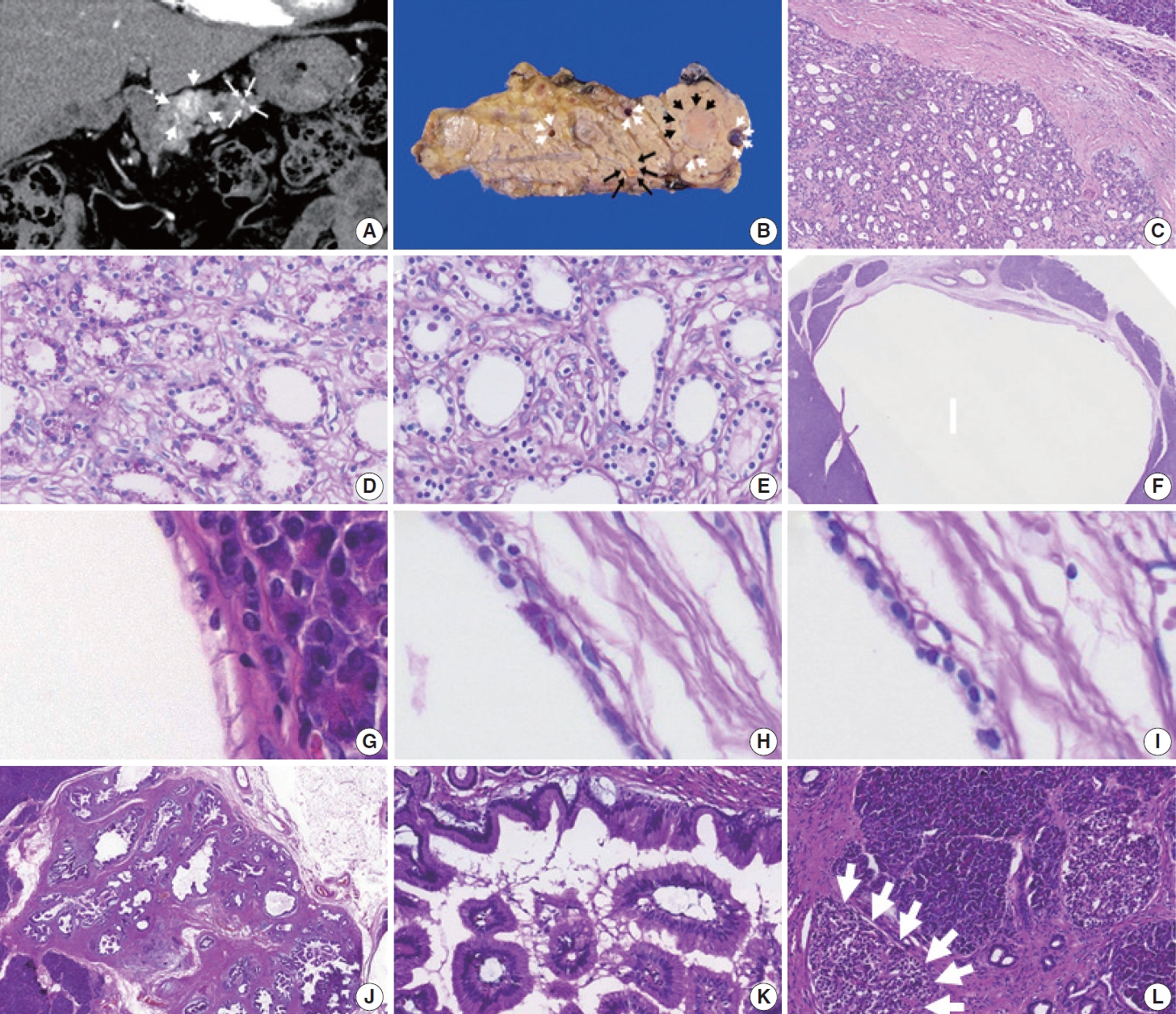
- At this visit, 10 years after the previous pancreatic resection for the PanNETs, an abdominal computed tomography scan revealed a 2 cm well-defined hypervascular mass and two minor sub-centimeter-sized hypervascular lesions in the proximal body of the remaining pancreas (Fig. 1A). The preoperative radiologic impression of the hypervascular lesions in the pancreatic body was either recurrent PanNETs or metastatic renal cell carcinomas. There were no demonstrable tumors in the stomach, duodenum, or residual kidneys. A completion pancreatectomy was then performed.
- A few solid masses and cysts were noted in the completion pancreatectomy specimen. The largest mass was a 2.2 cm, wellcircumscribed, yellow ovoid mass (black arrowheads; Fig. 1B) in the pancreatic body. The solid tumor had many tiny microcysts lined by monomorphic cuboidal cells with clear cytoplasm and bland oval nuclei in fibrotic stroma (Fig. 1C). The clear cytoplasm showed periodic acid-Schiff (PAS)–positivity (Fig. 1D), which was eliminated by diastase treatment, and these findings indicated that the tumor cells contained glycogen (Fig. 1E). Despite appearing solid upon gross examination, the tumor’s histologic features were consistent with a serous cystic neoplasm, and was diagnosed as a solid variant of serous cystic neoplasm.
- There were five unilocular cysts up to 0.9 cm in size (white arrowheads; Fig. 1B, F) adjacent to the solid variant of a serous cystic neoplasm. The cysts were also lined by non-mucinous cuboidal epithelial cells with clear cytoplasm and uniform, bland nuclei (Fig. 1G). The clear cytoplasm showed PAS-positivity (Fig. 1H), which was eliminated with diastase treatment (Fig. 1I). Therefore, the cystic tumors were diagnosed as a macrocystic variant of a serous cystic neoplasm.
- Additional gastric subtype low-grade intraductal papillary mucinous neoplasms with papillary projections involved the main and branch ductal lumens, measuring 1.6 cm in the greatest dimension (Fig. 1J). They were lined by tall columnar mucin-containing foveolar-type epithelial cells (Fig. 1K). Numerous islets with an increased size (up to 350 μm in the greatest dimension) were observed throughout the completion pancreatectomy specimen and were diagnosed as islet hyperplasia (Fig. 1L).
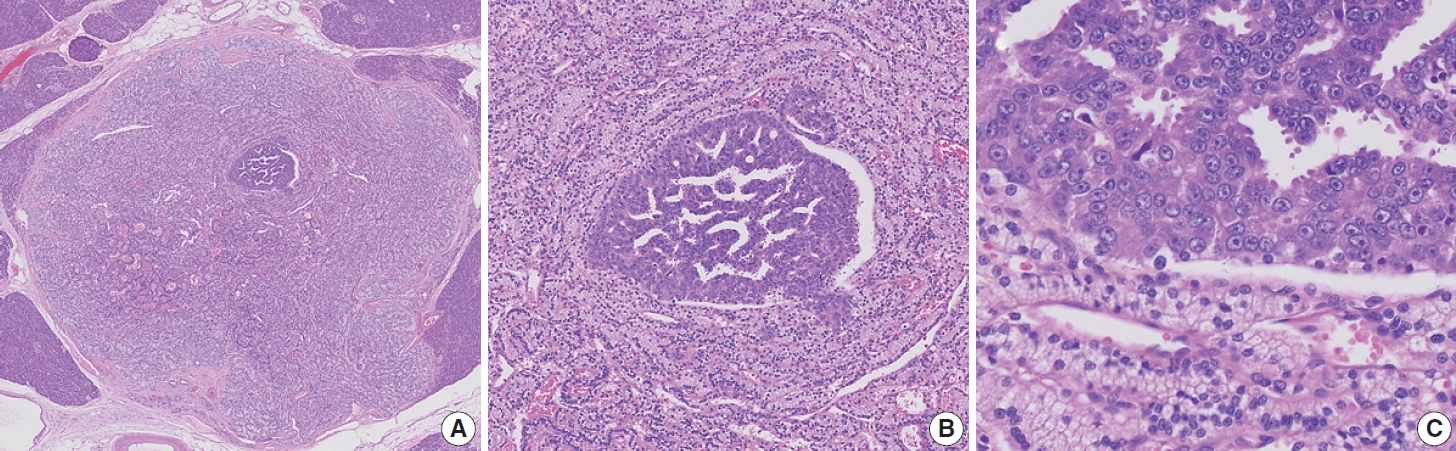
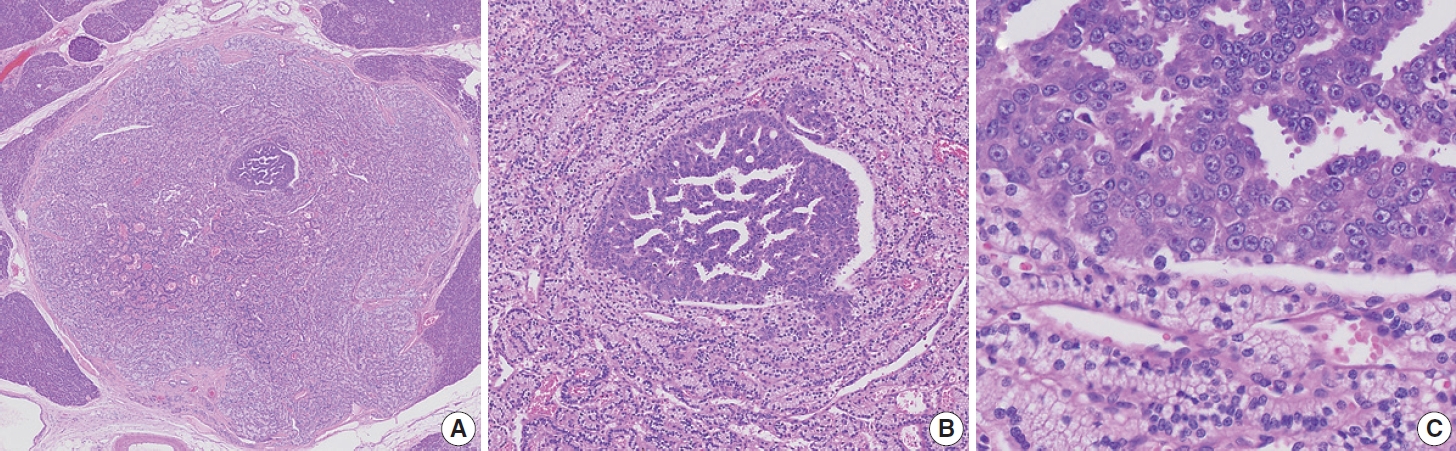
- Finally, a 0.4 cm, yellow, well-demarcated solid tumor with focal central red spots was observed in the pancreatic body (black arrows; Fig. 1B). The yellow tumor was cellular with a well-circumscribed nodule-in-nodule pattern (Fig. 2A). The outer nodule measured 0.4 cm in the greatest dimension and contained lightly staining monomorphic tumor cells with predominantly trabecular, focal pseudo-glandular patterns. The inner nodule was mostly circumscribed with a partial infiltration into the outer nodule (Fig. 2B). The outer nodule consisted of tumor cells with clear cytoplasm, round nuclei, and indistinct nucleoli at a higher power magnification (Fig 2C, upper half). The inner nodule, on the other hand, was 0.1 cm in size and contained darkly staining monomorphic tumor cells with a trabecular pattern (Fig 2C, lower half), and at a higher power magnification, amphophilic cytoplasm, vesicular nuclei, and prominent nucleoli were visible. Outside the tumor cells, capillaries were evenly distributed in both the inner and outer nodules. There was no geographic or spotty necrosis in either the inner or outer nodule. The mitotic counts of the tumor cells in the outer and inner nodules were 0/2 mm2 and 10/2 mm2, respectively.
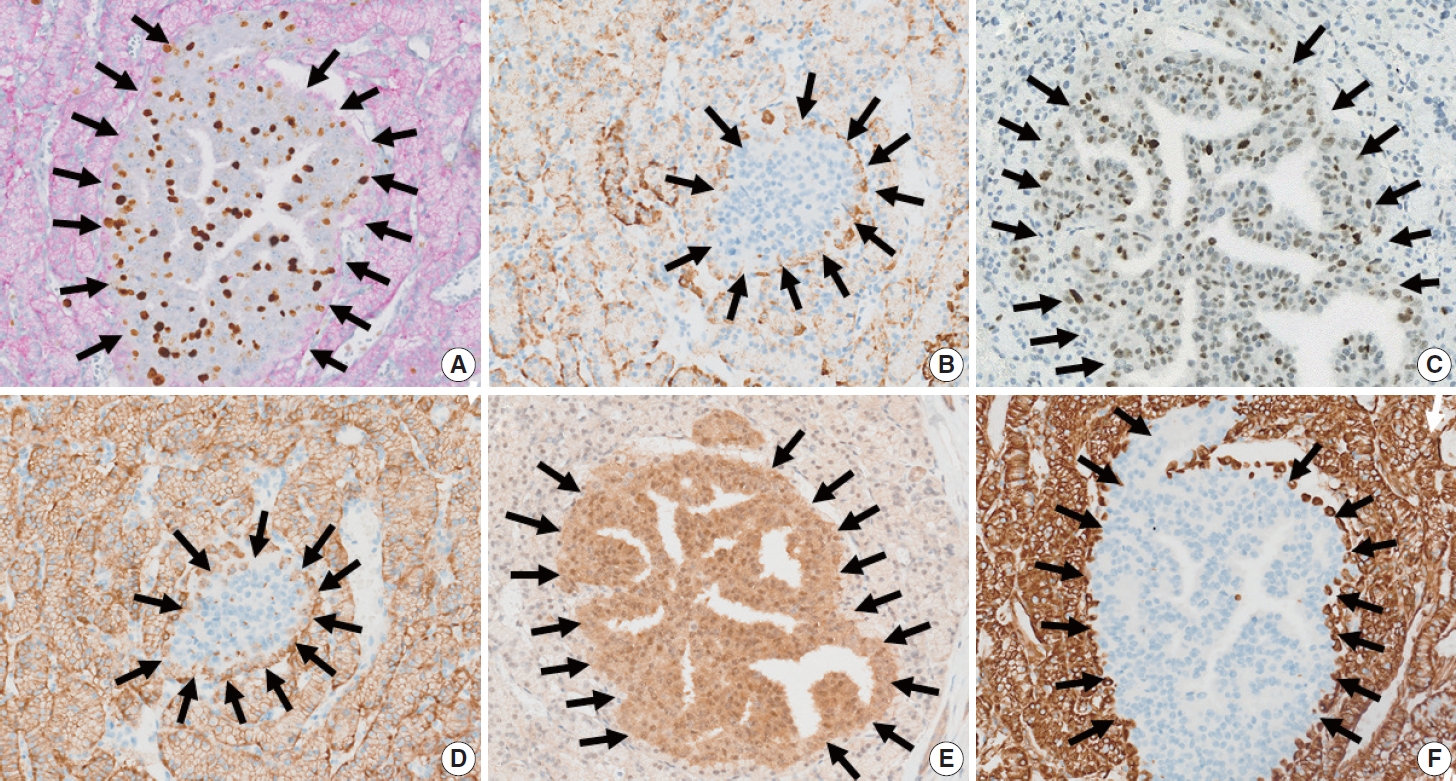
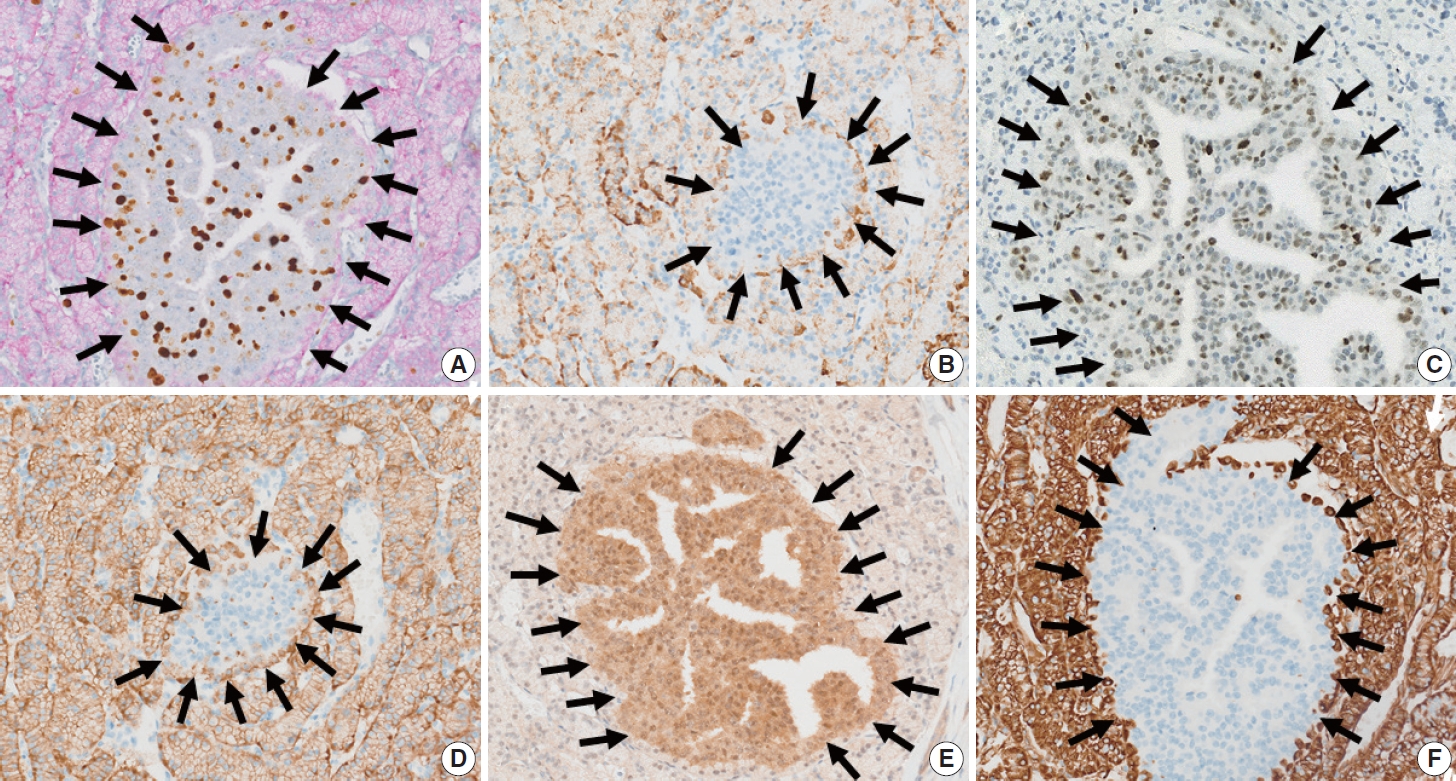
- The immunolabeling profiles of the neoplastic cells in the inner and outer nodules differed, as shown in Table 1. Double Ki-67 and synaptophysin immunolabeling showed diffuse synaptophysin positivity in the outer nodule, and focal rim positivity in the inner nodule (Fig. 3A). The Ki-67 labeling index (Ki-67 LI) of the outer and inner nodules was 2.1% (471/22,388 cells, grade 1) and 44.3% (140/316, grade 3), respectively by manual counting after image capture. Chromogranin immunolabeling revealed diffuse positivity in the outer nodule and focal positivity in the inner nodule (Fig. 3B). Tumor cells in the inner nodule were dif-fusely immunolabeled for p53 (Fig. 3C). In contrast, p53 expression was not observed in the outer nodule. Carbonic anhydrase 9 (CA9) immunolabeling was diffusely positive in the outer nodule, but was negative in the inner nodule (Fig. 3D). Smad4 immunolabeling was intact in the inner nodule but diminished in the outer nodule (Fig. 3E). Vimentin expression was observed in the outer nodule only (Fig. 3F). Tumor cells in both the inner and outer nodules expressed cytoplasmic retinoblastoma (Rb), nuclear ATRX, and DAXX immunolabelings. Both the inner and outer nodules were negative for Bcl-10. Weak insulin immunolabeling was observed diffusely in the inner nodule and focally in the outer nodule.
- The final pathologic diagnosis of the 0.4 cm yellow solid tumor was a PNEMT (grade 1) with a focal HGT (grade 3). As a result, the completion pancreatectomy specimen was diagnosed as a pancreas from a VHL syndrome patient containing solid and macrocystic variants of serous cystic neoplasms, as well as a concurrent grade 1 PNEMT with a focal grade 3 transformation.
- Follow-up information
- The patient has no evidence of recurrence or metastasis of the PNEMTs or PanNETs 36 months after the completion pancreatectomy. However, 4 months after the pancreatectomy, his serum prostate-specific antigen level increased. A 12-core prostate biopsy confirmed the presence of a prostate adenocarcinoma after several bone and lung metastases developed. He is currently receiving luteinizing hormone-releasing hormone agonist therapy.
- Immunohistochemical staining
- The entire surgically resected pancreas was submitted for evaluation, and 16 hematoxylin and eosin–stained sections were used. For the detection of glycogen, a PAS reaction with/without diastase treatment was used. The immunohistochemical labeling was performed at the Department of Pathology, Asan Medical Center. The primary antibodies were deployed with a Benchmark autostainer (Ventana Medical Systems, Tucson, AZ, USA) in accordance with the manufacturer’s protocol. Sections were incubated at room temperature for 32 minutes in primary antibodies against ATRX (1:300, Sigma-Aldrich, St. Louis, MO, USA), Bcl-10 (1:400, Santa Cruz Biotechnology, Santa Cruz, CA, USA), CA9 (1:100, Cell Marque, Rocklin, CA, USA), chromogranin (1:200, DakoCytomation, Glostrup, Denmark), cytokeratin 19 (1:100, Cell Marque), DAXX (1:100, Sigma-Aldrich), glucagon (1:200, Cell Marque), insulin (1:1,000, Novocastra, Newcastle upon Tyne, UK), Ki-67 (1:100 DakoCytomation), MUC-1 (1:200, Novocastra), PAX-8 (1:50, Cell Marque), p16 (1:6, Santa Cruz Biotechnology), p53 (1:1,000, DakoCytomation), Rb (1:10,000, QED Bioscience, San Diego, CA, USA), smad4 (1:1,000, Abcam, Cambridge, UK), synaptophysin (1:200, Cell Marque), and vimentin (1:500, Zymed, South San Francisco, CA, USA). The sections were subsequently processed using an iView DAB detection kit with an automated immunostaining system (Benchmark XT, Ventana Medical Systems) and counterstained with hematoxylin. Double Ki-67 and synaptophysin immunolabeling was performed as previously described [5]. The PNEMT was graded by the mitotic count and Ki-67 LI according to the WHO classification [1]; grade 1, < 2 mitoses/2 mm2; Ki-67 LI < 3%; grade 2, 2–20 mitoses/2 mm2 or a Ki-67 LI of 3%–20%; and grade 3, > 20 mitoses/2 mm2 or a Ki-67 LI > 20%.
CASE REPORT
- Pancreatic neuroendocrine microadenomas were previously considered biologically benign [1]. A recent report, however, suggested that a neuroendocrine microadenoma could be a malignant tumor because it metastasized to a peripancreatic lymph node [3]. Because of this metastatic potential, the term neuroendocrine microadenoma was changed to neuroendocrine microtumor in the 2022 WHO classification [4]. We present a rare case of a 0.4 cm PNEMT with focal HGT, which further substantiates our proposal that PNEMT are not always benign.
- The ‘nodule-in-nodule’ growth pattern is one of the steps in the multistep carcinogenesis model of hepatocellular carcinoma. The ‘nodule-in-nodule’ growth pattern manifests as a combination of an outer high-grade dysplastic nodule and an inner welldifferentiated hepatocellular carcinoma, or an outer well-differentiated hepatocellular carcinoma and an inner moderatelydifferentiated hepatocellular carcinoma [6]. The nodule-in-nodule pattern, on the other hand, has not previously been described for PanNETs or PNEMTs.
- In the current study, tumor cells in the inner nodule showed an infiltrative growth pattern. One recent study has reported a microscopic infiltrative tumor border in about 40% of surgically resected PanNETs, which was associated with higher tumor grades, larger tumors, more metastatic lymph nodes, higher American Joint Committee on Cancer stage groups, and the presence of lymphovascular and perineural invasions [7]. However, the clinicopathologic significance of a microscopic infiltrative tumor border in a PNEMT is currently unknown.
- As differential diagnoses for the inner nodule, several tumors, including metastatic clear cell type renal cell carcinomas, acinar cell carcinomas, and neuroendocrine carcinomas were considered. Metastatic clear cell type renal cell carcinoma was considered as one of the differential diagnoses because the patient had VHL syndrome and previously underwent a left partial nephrectomy for clear cell type renal cell carcinoma. Although tumor cells in the outer nodule had clear cytoplasm, PAX8 immunolabeling was negative. As a result, metastatic clear cell type renal cell carcinoma was ruled out. Acinar cell carcinomas and large cell type neuroendocrine carcinomas had to be ruled out because the tumor cells in the inner nodule had amphophilic cytoplasm and prominent nucleoli. Bcl-10 cytoplasmic immunolabeling was negative in the tumor cells of the inner nodule, and acinar cell carcinoma was excluded. The tumor cells in the inner nodule showed focal and weak rim synaptophysin positivity with increased Ki-67 LI (44.3%), consistent with a grade 3 PanNET or a poorly differentiated pancreatic neuroendocrine carcinoma (PanNEC). The tumor cells of the inner nodule, however had a trabecular growth pattern with evenly interspersed capillaries, which suggested a PanNET. Furthermore, a provisional panel of immunohistochemical staining supported by the literature, including p53, p16, MUC1, and smad4, was applied to differentiate between a grade 3 PanNET and a PanNEC [8], and the panel’s results favored a grade 3 PanNET over a PanNEC in the current case. In addition, chromogranin, CA9, and vimentin expression patterns differed between the inner and outer nodules. There was focal chromogranin labeling but no CA9 or vimentin immunolabeling tumor cells in the inner nodule. The outer nodule, conversely, showed diffuse chromogranin, CA9, and vimentin expression. Islets of Langerhans are vimentin-negative but are immunolabeled for CA9 [9]. CA9 and vimentin expressions were found in only 9% (2/23 cases) and 30% (7/23 cases) of PNEMTs, respectively, in a previous study [9]. In the same study, CA9 expression in PanNETs was associated with aggressive behavior and poor survival [9]. Vimentin expression has not been found to be related to the clinical behavior of PanNETs [9]. This may be evidence that CA9 participates in an early PanNET tumorigenesis stage, specifically, the microtumor stage, and is linked to a worse clinical outcome.
- PNEMTs can appear sporadically or as a part of hereditary syndromes [10-14]. The prevalence of PNEMTs varies with the amount of pancreatic tissue obtained (0.4%–10%) [15]. One previous autopsy study reported that the incidence of PNEMT was 1.6% (12/738) when three random histologic sections were submitted, but the incidence increased up to 10% (6/60) when entire pancreas tissues were submitted [2]. The most commonly reported grades of PNEMTs were grade 1, regardless of whether they were sporadic or syndromic in origin [12,16]. Only a few cases of grade 2 PNEMTs have previously been reported in the English-language literature [16,17]. De Wilde and colleagues observed one case of grade 2 microtumor out of 47 PNEMTs of the pancreas in patients with multiple endocrine neoplasia type 1 (MEN1) syndrome [17]. Furthermore, Okawa and colleagues reported one case of grade 2 PNEMT from a patient suspected of MEN1 syndrome among 23 PNEMTs evaluated for Ki67 LI (4%, 1 of 23) [16]. However, to the best of our knowledge, no PNEMT with histology greater than grade 2 has been reported in a patient with VHL syndrome in the English literature. The current case may be the first PNEMT containing distinct grade 1 and grade 3 areas with a nodule-in-nodule pattern. We did not define the area distinctions solely on the basis of KI-67 LI. Intratumoral differences in Ki-67 LI in large tumors, including Pan-NETs, could be interpreted as tumor heterogeneity [18]. However, distinct synaptophysin immunohistochemical staining patterns in inner and outer nodules, as seen in this case, cannot be explained by tumor heterogeneity alone.
- PanNETs are found in approximately 11% to 17% of VHL syndrome patients and are typically multiple tumors at the time of detection [19]. In addition to multiple PanNETs, patients with VHL syndrome may develop a number of precursor lesions, including PNEMTs [20]. Several histologic evidences of VHL syndrome were found in both the patient’s previous PPPD and the completion pancreatectomy specimens, including three PanNETs, one with grade 2 and two with grade 1 pathology, a PNEMT with HGT, multiple macrocystic serous cystic neoplasms, and a solid serous cystic neoplasm. Multiple neuroendocrine tumors that recur are uncommon in non-syndromic settings [19,21]. The current PanNETs have a clear cytoplasm, which has previously been reported as a feature of VHL syndrome [20]. Furthermore, the findings of multiple macrocystic serous cystic neoplasms in the PPPD and the completion pancreatectomy specimens are consistent with VHL syndrome involvement.
- As for study limitations, our study faced constraints in utilizing hypoxia-inducible factor 1α (HIF1α) and glucose transporter 1 (GLUT1) immunolabeling. The disappearance of the inner nodule in microtumor precluded the evaluation of the transformed area in the HIF1a and GLUT1 immunolabeling sections, impeding our ability to investigate these markers comprehensively.
- In summary, this case was diagnosed as a PNEMT with HGT. Our findings suggest that PNEMTs may not be benign tumors and can undergo HGT, as as exemplified by the current patient with VHL syndrome.
DISCUSSION
-
Ethics Statement
This case was deemed exempt from informed consent by the Asan Medical Center institutional review board (IRB #2022-0228).
-
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
-
Code Availability
Not applicable.
-
Author Contributions
Conceptualization: JL, SMH. Data curation: JL, KJL, DWH. Formal analysis: JL, KJL. Investigation: JL, SMH. Methodology: JL, SMH. Supervision: SMH. Validation: JL, SMH. Writing—original draft: JL, SMH. Writing—review & editing: JL, KJL, SMH. Approval of final manuscript: all authors.
-
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
-
Funding Statement
No funding to declare.
Notes
- 1. Nagtegaal ID, Odze RD, Klimstra D, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020; 76: 182–8. ArticlePubMedPMCPDF
- 2. Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci 1991; 36: 933–42. PubMed
- 3. Kwon JH, Kim HJ, Park DH, et al. Incidentally detected pancreatic neuroendocrine microadenoma with lymph node metastasis. Virchows Arch 2018; 473: 649–53. ArticlePubMedPDF
- 4. Rindi G, Mete O, Uccella S, et al. Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocr Pathol 2022; 33: 115–54. ArticlePubMedPDF
- 5. Ahn B, Jung JK, Jung H, et al. Double Ki-67 and synaptophysin labeling in pancreatic neuroendocrine tumor biopsies. Pancreatology 2022; 22: 427–34. ArticlePubMed
- 6. Kojiro M. ‘Nodule-in-nodule’ appearance in hepatocellular carcinoma: its significance as a morphologic marker of dedifferentiation. Intervirology 2004; 47: 179–83. ArticlePubMedPDF
- 7. Ahn B, Kim JY, Hong SM. Combined infiltrative macroscopic growth pattern and infiltrative microscopic tumor border status is a novel surrogate marker of poor prognosis in patients with pancreatic neuroendocrine tumor. Arch Pathol Lab Med 2023; 147: 100–16. ArticlePubMedPDF
- 8. Kim H, An S, Lee K, et al. Pancreatic high-grade neuroendocrine neoplasms in the Korean population: a multicenter study. Cancer Res Treat 2020; 52: 263–76. ArticlePubMedPMCPDF
- 9. Kim JY, Lee SH, An S, et al. Carbonic anhydrase 9 expression in well-differentiated pancreatic neuroendocrine neoplasms might be associated with aggressive behavior and poor survival. Virchows Arch 2018; 472: 739–48. ArticlePubMedPDF
- 10. Chetty R, Kennedy M, Ezzat S, Asa SL. Pancreatic endocrine pathology in von Hippel-Lindau disease: an expanding spectrum of lesions. Endocr Pathol 2004; 15: 141–8. ArticlePubMed
- 11. Chetty R, Ezzat S, Asa SL. Microadenomatosis of the pancreas in von Hippel-Lindau disease. Am J Surg Pathol 2006; 30: 1630.ArticlePubMed
- 12. Anlauf M, Schlenger R, Perren A, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol 2006; 30: 560–74. ArticlePubMed
- 13. Perigny M, Hammel P, Corcos O, et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: characterization by VHL/HIF pathway proteins expression. Am J Surg Pathol 2009; 33: 739–48. PubMed
- 14. Akatsu T, Aiura K, Ito Y, Ueda M, Kameyama K, Kitajima M. A novel Von Hippel-Lindau case with germline mutation at codon 167 (CGG to TGG) having endocrine microadenomatosis of the pancreas. Dig Dis Sci 2007; 52: 3145–8. ArticlePubMedPDF
- 15. Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system. Geneva: World Health Organization, 2010.
- 16. Okawa Y, Tsuchikawa T, Hatanaka KC, et al. Clinical features of pancreatic neuroendocrine microadenoma: a single-center experience and literature review. Pancreas 2022; 51: 338–44. ArticlePubMed
- 17. de Wilde RF, Heaphy CM, Maitra A, et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol 2012; 25: 1033–9. ArticlePubMedPMCPDF
- 18. Hwang HS, Kim Y, An S, et al. Grading by the Ki-67 labeling index of endoscopic ultrasound-guided fine needle aspiration biopsy specimens of pancreatic neuroendocrine tumors can be underestimated. Pancreas 2018; 47: 1296–303. ArticlePubMed
- 19. Lubensky IA, Pack S, Ault D, et al. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol 1998; 153: 223–31. PubMedPMC
- 20. Cassol C, Mete O. Endocrine manifestations of von Hippel-Lindau disease. Arch Pathol Lab Med 2015; 139: 263–8. ArticlePubMedPDF
- 21. Hammel PR, Vilgrain V, Terris B, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology 2000; 119: 1087–95. ArticlePubMed
References
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-