E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 54(5); 2020 > Article
-
Review
Immune landscape and biomarkers for immuno-oncology in colorectal cancers -
Jeong Mo Bae
 , Seung-Yeon Yoo, Jung Ho Kim, Gyeong Hoon Kang,
, Seung-Yeon Yoo, Jung Ho Kim, Gyeong Hoon Kang, -
Journal of Pathology and Translational Medicine 2020;54(5):351-360.
DOI: https://doi.org/10.4132/jptm.2020.05.15
Published online: June 26, 2020
Department of Pathology, Seoul National University College of Medicine, Seoul, Korea
- Corresponding Author: Gyeong Hoon Kang, MD, PhD, Department of Pathology, Seoul National University College of Medicine, 103 Daehak-ro, Jongno-gu, Seoul 03080, Korea Tel: +82-2-740-8263, Fax: +82-2-765-5600, E-mail: 'ghkang@snu.ac.kr'
© 2020 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Recent advances in immuno-oncology have increased understanding of the tumor immune microenvironment (TIME), and clinical trials for immune checkpoint inhibitor treatment have shown remission and/or durable response in certain proportions of patients stratified by predictive biomarkers. The TIME in colorectal cancer (CRC) was initially evaluated several decades ago. The prognostic value of the immune response to tumors, including tumor-infiltrating lymphocytes, peritumoral lymphoid reaction, and Crohn’s-like lymphoid reaction, has been well demonstrated. In this review, we describe the chronology of TIME research and review the up-to-date high-dimensional TIME landscape of CRC. We also summarize the clinical relevance of several biomarkers associated with immunotherapy in CRC, such as microsatellite instability, tumor mutational burden, POLE/POLD mutation, consensus molecular subtype, and programmed death-ligand 1 expression.
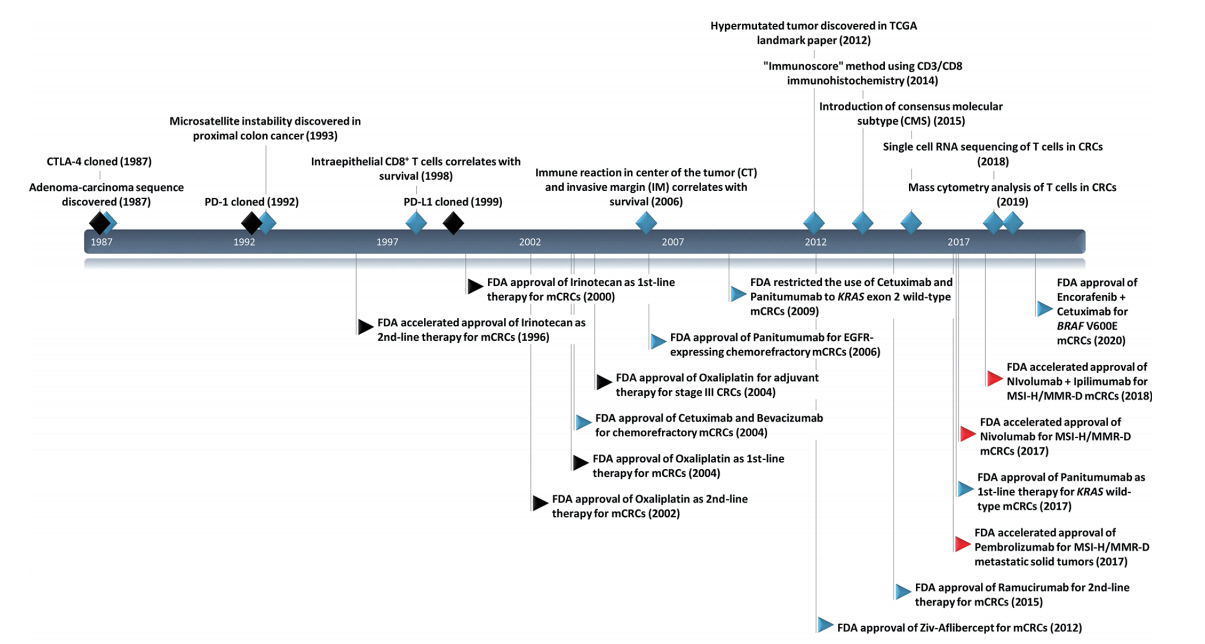
- Historical use of microscopic evaluation of immune environment in CRCs
- To our knowledge, Spratt and Spjut [9] published the first study on integrative histologic evaluation of CRCs in 1967. In that study, the authors evaluated histologic grade, mucinous elements, depth of invasion, characteristics of tumor border, lymphatic/vascular/perineural invasion, and especially degree of inflammatory reaction around the tumor in 1,137 consecutive CRC cases. CRC with no inflammatory reaction showed shorter 5-year and 10-year survivals than CRC with moderate inflammatory reaction and abscess formation. Watt and House [10] evaluated peritumoral lymphoid cells in a semiquantitative manner, and Duke B tumors with recurrence or death during follow-up showed a tendency toward minimal lymphocytic infiltration. Pihl et al. [11] reported the association of perivascular lymphoid cuffing in the muscularis propria or subserosa with favorable disease-free survival (DFS) in 134 Duke B CRCs. Perivascular lymphoid cuffing has been named “Crohn’s-like lymphoid reaction,” and is now referred to as an ectopic or tertiary lymphoid structure [12]. Naito et al. [13] evaluated intraepithelial tumor-infiltrating CD8+ T cells using immunohistochemistry and found that increased intraepithelial CD8+ cell infiltration was associated with lower Duke stage and better survival. Ogino et al. [14] reported that the overall lymphocytic reaction, which combines the degree of Crohn’s-like lymphoid reaction, peritumoral reaction, intratumoral periglandular reaction, and tumor-infiltrating lymphocytes (TILs), is a prognostic marker independent of clinicopathologic and molecular characteristics.
- Quantitative evaluation of TIME using digital pathology
- Previous studies evaluating the immune microenvironment in CRCs depended on manual inspection of glass slides using a light microscope. Visual assessment of immune cells and stroma is labor-intensive and has limited objectivity. Most studies enumerated immune cells in hotspot areas, while some studies used semiquantitative methods [15]. Recent advancements in virtual slide scanners and machine learning algorithms have enabled objective quantification of the tumor microenvironment at the whole slide level.
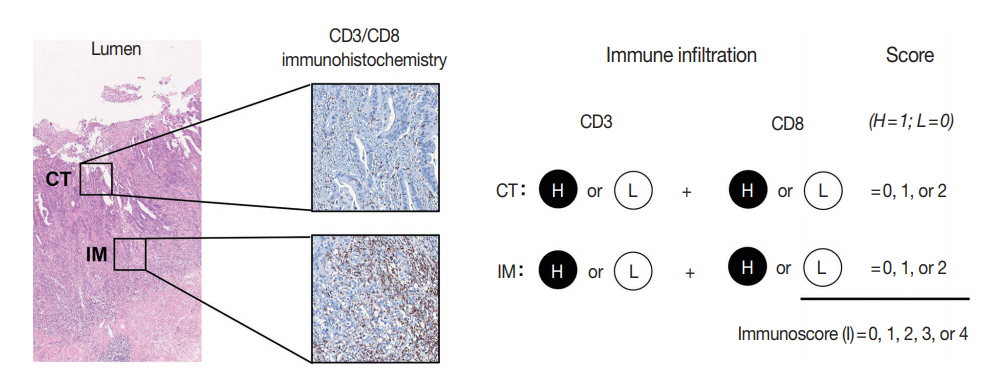
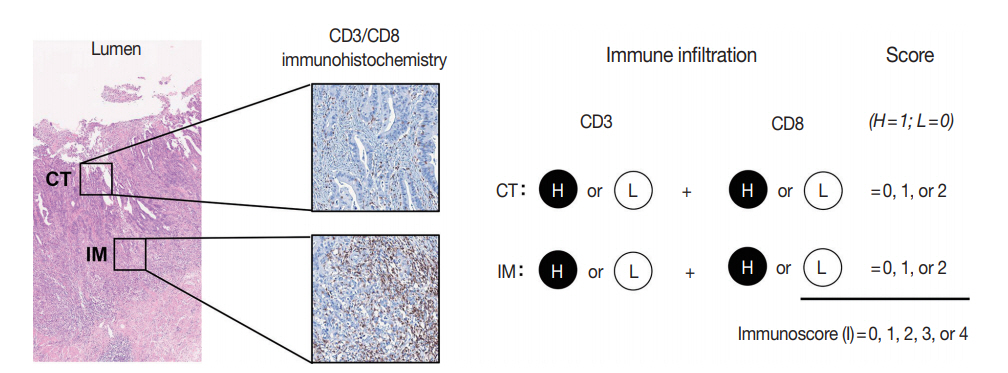
- The immunoscore developed by Jérôme Galon is the most well-known digital pathology-based approach for evaluation of TILs. In 2006, Galon et al. [16] showed that an increased number of total T cells (CD3+) and resident memory T cells (CD45RO+) in the center of the tumor (CT) and at the invasive margin (IM) was an independent marker of better DFS in CRCs. In 2014, they proposed the “Immunoscore” method based on enumeration of CD3+ and CD8+ T cells in the CT and IM regions of tumors (Fig. 2) [17]. Immunoscore assay shows superior prognostic value compared with microsatellite instability and has prognostic value in both primary tissues and metastatic tissues [18,19]. To ensure robust enumeration of TILs, the researchers developed an in vitro diagnostic immunoscore assay for clinical use. The international immunoscore consortium consisted of 14 institutions in 13 countries and performed a validation study for the immunoscore assay using 2,681 stage I–III colon cancers [20]. Using standardized immunohistochemistry protocols and image analysis software, the immunoscore assay showed high reproducibility between institutions. The consortium categorized CRC cases as low immunoscore group (0–25 percentile), intermediate immunoscore group (25–70 percentile), and high immunoscore group (70–100 percentile). Patients with high immunoscore showed longer time to recurrence compared to patients with low or intermediate immunoscore independent of age, sex, stage, microsatellite instability (MSI), and other known prognostic factors. Moreover, patients with high immunoscore showed significantly lower risk of recurrence compared to patients with low immunoscore in stage II colon cancers.
- Reichling et al. [21] performed whole-slide imaging analysis of 1,018 stage III colon cancers in the PETACC08 study. They developed software to detect colon cancer, normal mucosa, stroma, and immune cells on CD3- and CD8-stained slides. In their study, the stromal area in IM and CT (originally, tumor core in their article) showed strong positive correlation. A higher proportion of stromal area in the tumor was associated with poor relapse-free survival (RFS), and an increased proportion of stromal area in the IM showed the highest hazard ratio for RFS compared with the stromal area in the CT or total stromal area. The researchers tested the prognostic role of the four immune variables (mean values of each tumor tile for each slide), CD3+ IM, CD3+ CT, CD8+ IM, and CD8+ CT. High CD3+ IM, CD3+ CT, and CD8+ CT were significantly associated with superior clinical outcomes. The classical “Immunoscore” showed similar performance to the CD3+ CT variable in predicting clinical outcome. The researchers developed the “DGMate” score derived from 127 parameters extracted from image analysis and the “DGMuneS” score by combining CD3+ CT, stromal area in the IM, and “DGMate” score. The “DGMuneS” score slightly outperformed the “Immunoscore” in predicting clinical outcome.
- Nearchou et al. [15] evaluated tumor bud (TB), CD3+ TILs, and CD8+ TILs on the same section of stage II CRC tissues using multiplex immunofluorescence staining. They found that a higher density of CD3+ T cells at the IM and CD8+ T cells at the IM and in whole tumor sections (WTSs) were significantly correlated with lower TB number. In survival analysis using least absolute shrinkage and selection operator penalized Cox proportional hazard regression, four morphologic features, including CD3+ T cell density in WTSs, mean CD3+ CD8+ T cell number within 50 μm of TBs, TB number, and CD8+ T cell density in the CT, had significant predictive value for diseasespecific survival. After eliminating the least significant features, the authors proposed the “Tumor Bud-Immuno Spatial Index (TBISI)” using CD3+ T cell density in WTSs, mean CD3+ CD8+ T cell number within 50 μm of TBs, and TB numbers, which could predict disease-specific survival.
- We recently analyzed CD3+ TILs, CD8+ TILs, and the tumorstroma ratio (TSR) in 886 stage III or high-risk stage II CRCs using whole-slide imaging [22]. Clustering analysis using 197 parameters extracted from image analysis classified CRCs into five clusters. Strikingly, the five clusters showed similar clinicopathologic and molecular characteristics with the consensus molecular subtype (CMS) classification [23]. In detail, cluster 1 was characterized by highest TIL density, enrichment of MSI-H tumors, and CpG island methylator phenotype-high (CIMP-H) tumors (features of CMS1). Cluster 2 was characterized by lower TSR, distal colorectum locations, and retained intestinal differentiation (features of CMS2). Cluster 3 was characterized by the highest CD8/CD3 ratio and prominent mucin production (features of CMS3). Cluster 4 was characterized by the lowest TIL density and highest TSR (features of CMS4). Cluster 5 showed intermediate TIME characteristics, and this cluster was similar to tumors with mixed/indeterminate features in the original CMS study. This similarity highlights the clear association of the tumor transcriptome with the immune microenvironment [24]. Similar to the original CMS classification, cluster 4 in our study showed poor 5-year RFS in two independent datasets.
- High-dimensional analysis of immune landscape of CRCs
- Recent advancements in RNA sequencing, proteomics, and single-cell technologies have dramatically increased our understanding of the TIME. Deconvolution algorithms such as CIBERSORT [25], xCELL [26], and microenvironment cell populations-counter (MCP-counter) [27] for bulk RNA sequencing are useful tools for transcriptome data [28]. Because there are many publicly available transcriptome data for CRCs combined with genetic and clinicopathologic data from different datasets, TIME characteristics of each molecular or pathologic subtype can be identified. Using gene expression signatures, these algorithms can identify the cellular fractions of 6 to 64 immune and nonimmune cells. Mass cytometry provides high-dimensional protein-based cellular data for up to 40 antibodies at an individual cell level [29]. Combining cytometry and the time-of-flight method using lanthanide metal ion-tagged antibodies, mass cytometry provides high-dimensional data with low background noise. Single-cell RNA sequencing (scRNA-seq) provides unbiased profiling of immune cells without prior gene selection [30]. scRNA-seq enables classification of different subsets and identification of novel markers or regulators for each subset. Both mass cytometry and scRNA-seq used in an in-situ manner provide a more comprehensive TIME landscape by preserving spatial information [31,32].
- Xiong et al. [33] analyzed the proportion of 22 cell types in bulk transcriptome data from 2,306 patients with CRC (644 from The Cancer Genome Atlas [TCGA] RNA-sequencing data and 1,662 from Gene Expression Omnibus expression microarray data) using CIBERSORT. Tumor tissues showed more M0 and M1 macrophages, resting natural killer (NK) cells, plasma cells, and memory and activated CD4+ T cells along with fewer resting mast cells and M2 macrophages than normal tissues. In survival analysis, M1 macrophages and activated dendritic cells were significantly associated with improved outcome, whereas eosinophils, neutrophils, and M2 macrophages were associated with poorer outcomes. Marisa et al. [34] quantified immune cell infiltration in transcriptome data using an MCP-counter. There was a strong positive association between immune checkpoint expression with infiltration of certain lymphoid (NK cells, T cells, and cytotoxic cells) and myeloid cells, whereas B cells, fibroblasts, vessels, and granulocytes showed little or no association with immune checkpoint expression.
- Zhang et al. [35] performed scRNA-seq and T cell receptor (TCR) tracking to analyze distinct functions and clonalities among 11,138 T cells from 12 patients with CRC. Using the tstochastic neighbor embedding method, the authors found a total of eight CD8+ and 12 CD4+ T cell clusters. Within CD8+ T cells, naïve T cells, central memory T cells, and recently activated effector memory T (TEMRA) cells were enriched in blood, whereas exhausted T (TEX) cells were specifically enriched in tumors. Resident memory T cells were predominantly found in normal mucosa. Among CD4+ subtypes, naïve and effector-like cells were enriched in blood. Follicular helper T cells were enriched in normal mucosa, whereas two IFNG+ Th1-cell-like subsets and Th17 cells were enriched in tumors. In clonality analysis, CD8+ TEX cells and TEMRA cells showed the highest degree of clonal expansion. Among CD4+ T cells, most tumor-infiltrating regulatory T (Treg) cells clones showed clonal exclusivity, whereas certain Treg cell clones were developmentally linked to several helper T cell clones. The researchers also found that CXCL13+ BHLHE40+ Th1-like cells were abundant in MSI tumors, whereas microsatellite-stable (MSS) tumors were moderately enriched with TH17 cells. BHLHE40 is expressed in T cells via TCR stimulation, which positively regulates granulocyte-macrophage colony-stimulating factor and IFN-γ production [36,37]. The authors speculated that enrichment of CXCL13+ BHLHE40+ IFNG+ Th1-like cells might be one cause of a favorable response to immunotherapy in patients with MSI CRC. Recently, anti-CD40 agonist treatment was reported to increase BHLHE40+ Th1-like cells in a MC38 syngeneic mouse tumor model. This finding suggests crosstalk of tumor-associated BHLHE40+ Th1-like cells and conventional type 1 dendritic cells [38].
- De Vries et al. [39] performed single-cell mass cytometry using 36 immune cell markers in 35 CRC tissues, 26 tumor-associated lymph nodes, 17 healthy mucosae, and 19 peripheral blood samples from 31 patients with CRC. Clustering analysis of CD8+/γδ T cells revealed that activated (HLA-DR+ CD38+ PD-l+) and tissue-resident (CD103+ CD69+) phenotypes were enriched in tumor tissue compared with other tissues. Clustering analysis of CD4+ T cells showed that inducible T-cell co-stimulator (ICOS)+ CD27– cells were enriched in tumor tissues, and these cells showed a regulatory-like phenotype overexpressing FOXP3. In innate lymphocyte populations, Lin– CD7+ CD127– CD56+ CD45RO+ cells were enriched in tumor tissues, accounting for up to 80% of the innate lymphoid compartment. This subset showed a tissue-resident (CD103+ CD69+) phenotype and displayed cytotoxic activity. Moreover, this cell population was abundant in mismatch repair (MMR)-deficient tumors. Norton et al. [40] showed that B lymphocyte-induced maturation protein-1 (BLIMP1)+ Treg cells were significantly enriched in tumor tissues compared with normal mucosa. The enrichment of ICOS, CD45RO, PD-1, programmed death-ligand 1 (PD-L1), lymphocyte-activation gene 3 (LAG-3), CTLA-4, and T-cell immunoglobulin mucin-3 (TIM-3) on BLIMP-1+ regulatory T cells suggested that BLIMP-1+ Treg cells have a more activated phenotype than conventional Treg cells and may play a role in the antitumor immune response. Di et al. [41] also found that exhausted T cells (PD-1+ CD38+ HLA-DR+ CCR7+ CD127–) and regulatory T cells (CD4+ CD25+ CD127–) were increased in tumor tissues. Moreover, they found that CD8+ CD28– immunosenescent T cells with impaired proliferation capacity were the most abundant T cell population in colorectal tumors.
IMMUNE LANDSCAPE OF COLORECTAL CANCERS
- Microsatellite instability/mismatch repair-deficiency
- Germline mutation of genes encoding MMR enzymes (MLH1, MSH2, MSH6, and PMS2) or promoter hypermethylation of the hMLH1 gene causes MMR deficiency (MMR-D) [42]. MMR-D causes numerous frameshift mutations that result in increased neoantigen production [43]. Increased neoantigen production causes vigorous immune reactions in MSI-H CRCs, such as increased TIL infiltration, peritumoral lymphocytic infiltration, and Crohn’s-like lymphoid reactions. How MSI-H CRCs persist in a hostile immune microenvironment is of great interest. Llosa et al. [44] showed that although MSI-H CRCs showed high infiltration by CD8+ cytotoxic T lymphocytes and Th1 cells, these tumors showed upregulated expression of immune checkpoint molecules, including PD-1, PD-L1, CTLA-4, LAG3, and indoleamine 2,3-dioxygenase. This finding suggested that MSI-H CRC may be a good candidate for ICI treatment. MSI-H/MMR-D CRCs are more responsive to PD-1 blockade than MSS/MMR-proficient CRCs [7]. Because MSI/MMR status showed predictive value in other extracolonic cancers, MSI/MMR status became a tissue-agnostic predictive biomarker for ICI treatment [45]. The reported response rate is 28%–52%, and the disease control rate is 51%–82% for ICI in MSI-H/MMR-D CRCs [7,8,46]. Currently, the anti-PD-1 inhibitor pembrolizumab can be used as third-line therapy for MSI-H/MMR-D CRCs by off-label use after agreement by a multi-disciplinary team in designated institutions in Korea.
- Tumor mutational burden
- Tumor mutational burden (TMB) is a measure of the total amount of somatic coding mutations in a tumor, and it is considered an emerging biomarker for ICI treatment [47]. Initially, the concept of TMB was derived from whole exome sequencing (WES); however, many studies revealed that TMB calculated from targeted next generation sequencing panels showed clinically compatible results with WES [48]. TMB values should be interpreted cautiously because the calculation formula for TMB varies among different panels, and the cut-off for TMB-high (TMB-H) status differs among clinical trials and tumor types [49].
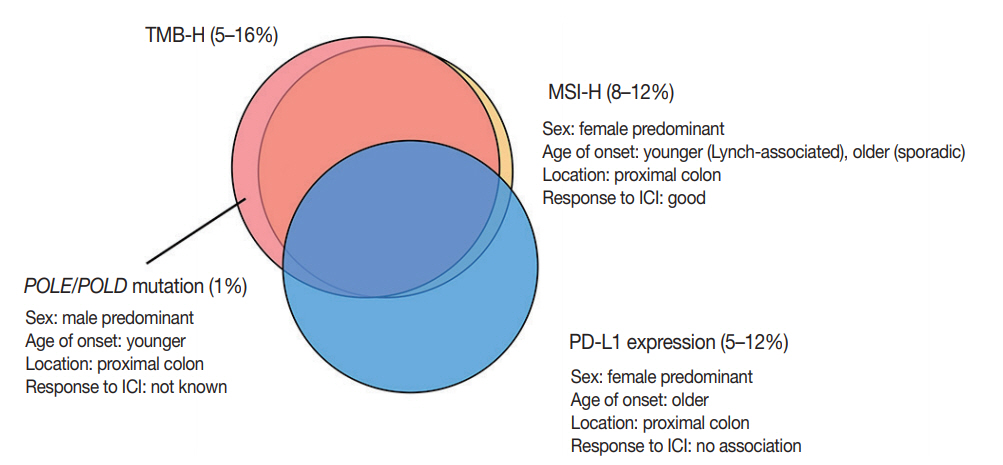
- TCGA consortium reported that 15.9% of 549 CRCs showed hypermutation (≥10/Mb) using WES [50]. However, Parikh et al. [51] reported that TMB-H was observed in 4.9% of 12,569 CRCs using a cut-off of ≥20/Mb analyzed by the FoundationOne panel. Although MSI is a proven predictive marker for ICI treatment, patients can be stratified further by TMB status. Schrock et al. [46] recently reported that patients showing objective response had higher TMB (median, 54/Mb; range, 31 to 91/Mb) than non-responders (median, 29/Mb; range, 13 to 37/ Mb) in 22 patients with metastatic MSI-H CRC treated with anti–PD-1 or anti–PD-L1 treatment [46]. Lee et al. [52] reported that TMB was a prognostic marker of better RFS in stage III or high-risk stage II CRCs treated with oxaliplatin-based adjuvant chemotherapy, independent of MSI status.
- Three-quarters of TMB-H CRCs are MSI-H, and the remaining one-quarter are MSS with somatic mutations in proofreading genes, mainly polymerase ε (POLE) and polymerase δ (POLD) [53]. In mutation analysis, MSI-H tumors show an insertiondeletion (indel)-predominant pattern, while POLE mutants show a single nucleotide variation–predominant pattern [54,55]. Domingo et al. [56] analyzed the frequency of somatic POLE mutations in 6,517 CRCs, and POLE mutations were detected in 1% of CRCs. CRCs with POLE mutations are associated with younger age at diagnosis, male sex, and proximal location [55,56]. Both POLE mutation and MSI-H/MMR-D status were associated with reduced risk of recurrence in a retrospective study [56].
- There are limited data about the predictive value of POLE mutations in ICI treatment. In Wang et al.’s study [57], three patients harboring pathogenic POLE mutations received ICI treatment, and only one patient showed complete remission. Silberman et al. [58] reported a case of complete and sustained response to anti–PD-1 treatment in a patient with metastatic CRCs harboring a pathogenic p.Val411Leu POLE mutation.
- Consensus molecular subtype
- Several investigators have suggested gene expression-based CRC classifications [59-61]. Due to the similarity among different classifications, the international CRC Subtyping Consortium proposed a unified transcriptomic classification, which was named the “consensus molecular subtype” (CMS) classification [23]. CMS1 (14%, MSI immune subtype) is characterized by MSI-H and CIMP-H statuses in addition to hypermutation and BRAF mutation. CMS1 also shows high immune infiltration and activation. CMS2 (37%, canonical subtype) is characterized by somatic copy number alterations and WNT and MYC activation. CMS3 (13%, metabolic subtype) shows metabolic deregulation and is associated with KRAS mutations. CMS4 (23%, mesenchymal subtype) is characterized by stromal infiltration, transforming growth factor β (TGF-β) activation, and angiogenesis. In the original CMS paper, CMS1 was associated with worse survival after relapse, while CMS4 was associated with worse RFS and overall survival.
- By applying deconvolution algorithms to bulk transcriptome data, Becht et al. [24] and Karpinski et al. [62] evaluated the cellular composition of each CMS subtype in CRCs. In Becht et al.’s study [24] using an MCP-counter, CMS1 and CMS4 showed high expression of lymphoid and myeloid cell-specific genes. However, CMS1 exhibited high infiltration by CD8+ cytotoxic T cells and NK cells, whereas CMS4 showed high expression of fibroblastic and endothelial cells [24]. Karpinski et al. [62] measured the proportion of 22 immune cell subtypes in 1,597 CRCs using CIBERSORT. CMS1 showed enrichment of leukocytes related to adaptive immunity (follicular helper T cells, memory activated helper T cells, and cytotoxic T cells) and innate immunity (activated NK cells, γδ T cells, M1 macrophages, activated dendritic cells, activated mast cells, and neutrophils). In addition, CMS1 showed depletion of Treg cells. CMS2 showed enrichment of helper T cells and memory B cells. CMS3 showed low levels of immune activation that manifested as high levels of resting memory helper T cells, naïve B cells, and low levels of macrophages, neutrophils, and activated helper T cells. Last, CMS4 was characterized by the highest proportions of leukocytes related to protumor activity (eosinophils, monocytes, M2 macrophages, resting dendritic cells, and regulatory T cells).
- Soldevilla et al. [63] evaluated the distribution of six immune subtypes proposed by Thorsson et al. [64] among each CMS subtype in TCGA samples. In a total of 625 samples, the C1 wound healing subtype (77%) was the most predominant, followed by the C2 INF-γ dominant subtype (17%). The C1 wound healing subtype was predominant in the CMS2 subtype (92%) but less common in the CMS1 subtype (46%). In contrast, the C2 IFN-γ dominant subtype was the most common immune subtype in CMS1 (53%) and was underrepresented in CMS2 (8%). CMS3 showed a higher frequency of the C3 inflammatory subtype (7%) and the C4 lymphocyte-depleted subtype (4%) than the other CMSs. The C6 TGF-β dominant subtype was exclusively observed in CMS4.
- Marisa et al. [34] evaluated whether the prognostic value of immune gene expression varies according to CMS. The expression of genes associated with Th1 cells, cytotoxic T cells, and cytotoxicity was predictive of better prognosis in MSS tumors from CMS2 and CMS3. In contrast, they had no prognostic relevance in CMS1 and CMS4. A brief summary of genomic and immunologic characteristics of each CMS subtype is shown in Table 1.
- PD-L1 expression
- Several studies evaluated the clinicopathologic characteristics of PD-L1 expression in CRCs. The frequency of PD-L1 overexpression in tumor cells is approximately 5% to 12% in all CRCs (1 to 4% in MSS CRCs and 18% to 45% in MSI-H CRCs) [65-69]. PD-L1 overexpression is associated with female sex, right-sided colon occurrence, poor differentiation, solid or medullary histology, increased number of TILs, and BRAF V600E mutation. Although a recent meta-analysis showed that PD-L1 overexpression in tumor cells was significantly associated with poor overall survival and decreased DFS, the prognostic value of PD-L1 overexpression differed according to MSI status [70]. Lee et al. [66] showed that the association of decreased recurrence-free survival with PD-L1 overexpression was only observed in MSI-H CRCs. In Marisa et al.’s transcriptome analysis [34], there was a strong association of immune checkpoint expression with infiltration of specific lymphoid (NK cells, T cells, and cytotoxic cells) and myeloid cells, whereas B cells, fibroblasts, vessels, and granulocytes showed little or no association with immune checkpoint expression. Patients with MSI-H CRC with high immune checkpoint expression, including high expression of PD-L1, showed significantly poorer overall survival compared to patients with MSI-H CRC and low immune checkpoint expression. However, immune checkpoint expression status did not influence the clinical outcome of patients with MSS CRC.
- In addition, the frequency of PD-L1 expression in MSI-H CRCs differs by the etiology of MSI. In our previous study, MSI CRCs with MLH1 methylation showed a higher frequency of PD-L1 expression (28.3%) than MSI CRCs without MLH1 methylation (6.1%) [67]. Yamada et al. [71] reported similar results, which showed increased PD-L1 expression in sporadic MSI-H CRCs (25.0%) compared to Lynch syndrome-associated CRCs (3.6%). The underlying mechanism of PD-L1 overexpression in sporadic MSI CRCs should be further investigated.
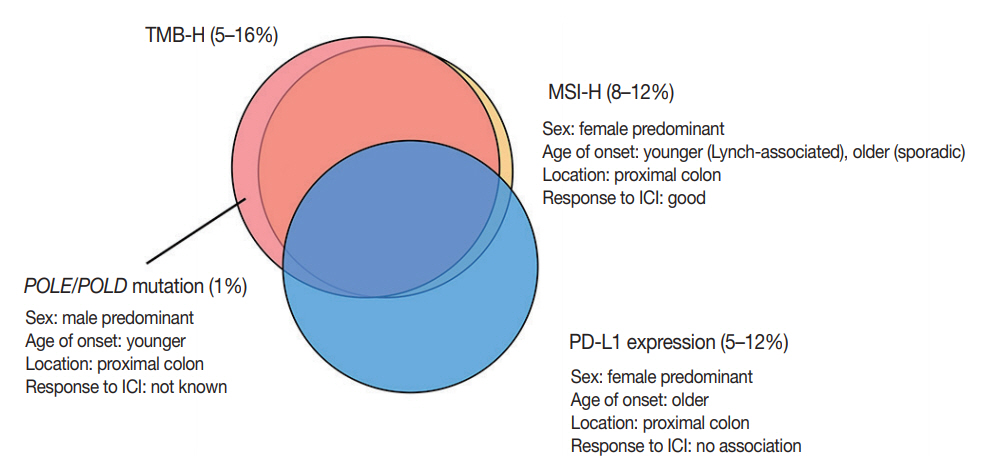
- Although PD-L1 expression is considered a predictive marker for ICI treatment in several types of cancer, such as melanoma and non-small-cell lung cancer, currently available data have demonstrated that PD-L1 expression has no predictive value in CRC with ICI treatment [7,8]. The discrepancy between PDL1 expression and response to ICI treatment might be related to spatiotemporal variations in PD-L1 expression, the cut-off for PD-L1 positivity, or interobserver variations in interpretations. The correlations of each biomarker described above are summarized in Fig. 3.
BIOMARKERS ASSOCIATED WITH IMMUNE MICROENVIRONMENT AND IMMUNOTHERAPY
- Evaluation of the TIME in CRCs has advanced from semiquantitative visual inspection to quantitative and high-dimensional approaches. Numerous clinical trials are ongoing to develop next-generation immune checkpoint drugs that target checkpoint modulators beyond PD-1/PD-L1, such as TIM-3, LAG-3, and T cell immunoreceptor with Ig and ITIM domains [72]. Combination immunotherapies, which combine cytotoxic chemotherapeutic agents or vascular endothelial growth factor inhibitors with ICIs, are also being investigated. The importance of comprehensive understanding of the TIME is constantly increasing in the era of immunotherapy. Moreover, numerous efforts are required to develop more precise biomarkers to classify responders to immunotherapy in CRC.
CONCLUSION
-
Ethics Statement
Not applicable.
-
Author contributions
Conceptualization: JMB, GHK. Funding acquisition: JMB. Investigation: JMB, SYY, JHK. Methodology: JMB, SYY, JHK. Supervision: GHK. Writing—original draft: JMB. Writing—review & editing: JMB, GHK. Approval of final manuscript: all authors.
-
Conflicts of Interest
J.H.K. and G.H.K., contributing editors of the Journal of Pathology and Translational Medicine, were not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
-
Funding
This study was supported by grants from the National Research Foundation (NRF) (Grant number: NRF-2017R1D1A1B03030073) and the Seoul National University Hospital (SNUH) Research Fund (Grant number: 04-2019-0560).
Notes
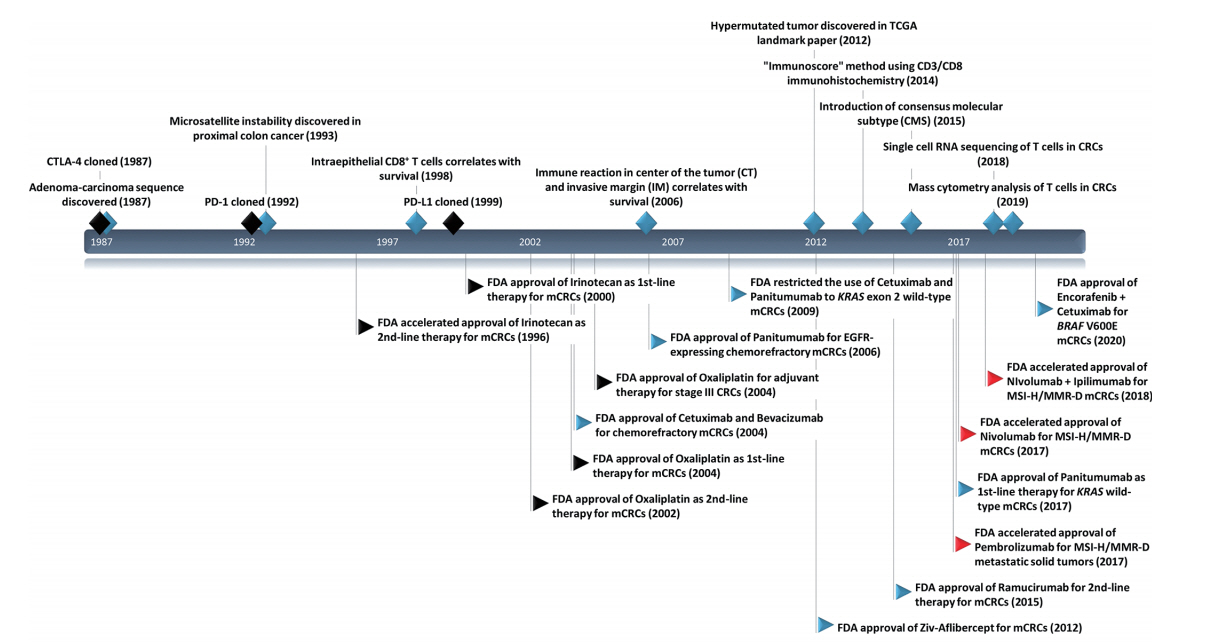
| CMS1 | CMS2 | CMS3 | CMS4 | |
|---|---|---|---|---|
| MSI immune (14%) | canonical (37%) | metabolic (13%) | mesenchymal (23%) | |
| Genomic feature | MSI | MSS | MSS | MSS |
| CIMP-positive | CIMP-negative | CIMP-low | CIMP-negative | |
| Hypermutation | SCNA-high | SCNA-intermediate | SCNA-high | |
| SCNA-low | ||||
| Expression signature | Immune infiltration and activation | WNT and MYC activation | Metabolic deregulatio | Stromal infiltration, TGF-β activation, angiogenesis |
| Immune-subtype | Immune activated | Immune desert | Immune excluded | Immune inflamed |
| Immune cell infiltration | CD8+ TIL ↑ | MHC1 (HLA, B2M) ↓ | MDSC↑ | |
| Macrophages ↑ | Treg ↑ | |||
| NK cells ↑ | Th17 ↑ | |||
| Immune gene expression | T cell chemotaxis and activation (CXCL9, CXCL10, CXCL16, IFN-γ) ↑ | Angiogenesis (VEGF) ↑ | ||
| T cell-specific inhibition (PD-1, PD-L1, CTLA-4, LAG-3) ↑ | Immunosuppression (TGF-β) ↑ | |||
| MHC1 (HLA, B2M) ↑ | Complement ↑ | |||
| Immune subtype by Thorsson et al. [63,64] | C2 INF-γ dominant ↑↑ | C1 wound healing ↑↑ | C3 inflammatory ↑ | C6 TGF-β dominant ↑ |
| C4 lymphocyte depleted ↑ | ||||
| TIME cluster by Yoo et al. [22] | Cluster 1, TIL ↑↑ | Cluster 2, TSR ↓↓ | Cluster 3, CD8/CD3 ↑↑ | Cluster 4, TSR ↑↑ |
CMS, consensus molecular subtype; MSI, microsatellite instability; MSS, microsatellite stable; CIMP, CpG island methylator phenotype; SCNA, somatic copy number alteration; TIL, tumor-infiltrating lymphocytes; NK, natural killer; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell; IFN, interferon; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte antigen 4; LAG-3, lymphocyte-activation gene 3; TGF-β, transforming growth factor-β;TSR, tumor-stroma ratio.
- 1. Jung KW, Won YJ, Kong HJ, Lee ES. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2016. Cancer Res Treat 2019; 51: 417–30. ArticlePubMedPMCPDF
- 2. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017; 67: 177–93. ArticlePubMed
- 3. Bae JM, Kim JH, Kwak Y, et al. Distinct clinical outcomes of two CIMP-positive colorectal cancer subtypes based on a revised CIMP classification system. Br J Cancer 2017; 116: 1012–20. ArticlePubMedPMCPDF
- 4. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017; 14: 717–34. ArticlePubMedPDF
- 5. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–23. ArticlePubMedPMC
- 6. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015; 372: 2018–28. ArticlePubMed
- 7. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372: 2509–20. ArticlePubMedPMC
- 8. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017; 18: 1182–91. ArticlePubMedPMC
- 9. Spratt JS Jr, Spjut HJ. Prevalence and prognosis of individual clinical and pathologic variables associated with colorectal carcinoma. Cancer 1967; 20: 1976–85. ArticlePubMed
- 10. Watt AG, House AK. Colonic carcinoma: a quantitative assessment of lymphocyte infiltration at the periphery of colonic tumors related to prognosis. Cancer 1978; 41: 279–82. ArticlePubMed
- 11. Pihl E, Malahy MA, Khankhanian N, Hersh EM, Mavligit GM. Immunomorphological features of prognostic significance in Dukes' Class B colorectal carcinoma. Cancer Res 1977; 37: 4145–9. PubMed
- 12. Maoz A, Dennis M, Greenson JK. The Crohn's-like lymphoid reaction to colorectal cancer-tertiary lymphoid structures with immunologic and potentially therapeutic relevance in colorectal cancer. Front Immunol 2019; 10: 1884.ArticlePubMedPMC
- 13. Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 1998; 58: 3491–4. PubMed
- 14. Ogino S, Nosho K, Irahara N, et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res 2009; 15: 6412–20. ArticlePubMedPMC
- 15. Nearchou IP, Lillard K, Gavriel CG, Ueno H, Harrison DJ, Caie PD. Automated analysis of lymphocytic infiltration, tumor budding, and their spatial relationship improves prognostic accuracy in colorectal cancer. Cancer Immunol Res 2019; 7: 609–20. ArticlePubMed
- 16. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–4. ArticlePubMed
- 17. Anitei MG, Zeitoun G, Mlecnik B, et al. Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin Cancer Res 2014; 20: 1891–9. ArticlePubMed
- 18. Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016; 44: 698–711. ArticlePubMed
- 19. Mlecnik B, Van den Eynde M, Bindea G, et al. Comprehensive intrametastatic immune quantification and major impact of immunoscore on survival. J Natl Cancer Inst 2018; 110: 97–108. ArticlePDF
- 20. Pages F, Mlecnik B, Marliot F, et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018; 391: 2128–39. ArticlePubMed
- 21. Reichling C, Taieb J, Derangere V, et al. Artificial intelligence-guided tissue analysis combined with immune infiltrate assessment predicts stage III colon cancer outcomes in PETACC08 study. Gut 2020; 69: 681–90. ArticlePubMed
- 22. Yoo SY, Park HE, Kim JH, et al. Whole-slide image analysis reveals quantitative landscape of tumor-immune microenvironment in colorectal cancers. Clin Cancer Res 2020; 26: 870–81. ArticlePubMed
- 23. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21: 1350–6. ArticlePubMedPMCPDF
- 24. Becht E, de Reynies A, Giraldo NA, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res 2016; 22: 4057–66. ArticlePubMed
- 25. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015; 12: 453–7. ArticlePubMedPMCPDF
- 26. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017; 18: 220.ArticlePubMedPMCPDF
- 27. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016; 17: 218.ArticlePubMedPMCPDF
- 28. Jimenez-Sanchez A, Cast O, Miller ML. Comprehensive benchmarking and integration of tumor microenvironment cell estimation methods. Cancer Res 2019; 79: 6238–46. ArticlePubMed
- 29. Labib M, Kelley SO. Single-cell analysis targeting the proteome. Nat Rev Chem 2020; 4: 143–58. ArticlePubMedPDF
- 30. Zhang L, Zhang Z. Recharacterizing tumor-infiltrating lymphocytes by single-cell RNA sequencing. Cancer Immunol Res 2019; 7: 1040–6. ArticlePubMed
- 31. Moncada R, Barkley D, Wagner F, et al. Integrating microarraybased spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol 2020; 38: 333–42. ArticlePubMedPDF
- 32. Jackson HW, Fischer JR, Zanotelli VR, et al. The single-cell pathology landscape of breast cancer. Nature 2020; 578: 615–20. ArticlePubMedPDF
- 33. Xiong Y, Wang K, Zhou H, Peng L, You W, Fu Z. Profiles of immune infiltration in colorectal cancer and their clinical significant: a gene expression-based study. Cancer Med 2018; 7: 4496–508. ArticlePubMedPMC
- 34. Marisa L, Svrcek M, Collura A, et al. The balance between cytotoxic T-cell lymphocytes and immune checkpoint expression in the prognosis of colon tumors. J Natl Cancer Inst 2018; 110: 68–77. ArticlePDF
- 35. Zhang L, Yu X, Zheng L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018; 564: 268–72. ArticlePubMedPDF
- 36. Miyazaki K, Miyazaki M, Guo Y, et al. The role of the basic helixloop-helix transcription factor Dec1 in the regulatory T cells. J Immunol 2010; 185: 7330–9. ArticlePubMed
- 37. Lin CC, Bradstreet TR, Schwarzkopf EA, et al. Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat Commun 2014; 5: 3551.ArticlePubMedPMCPDF
- 38. Zhang L, Li Z, Skrzypczynska KM, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 2020; 181: 442–59. ArticlePubMed
- 39. de Vries NL, van Unen V, Ijsselsteijn ME, et al. High-dimensional cytometric analysis of colorectal cancer reveals novel mediators of antitumour immunity. Gut 2020; 69: 691–703. ArticlePubMed
- 40. Norton SE, Ward-Hartstonge KA, McCall JL, et al. High-dimensional mass cytometric analysis reveals an increase in effector regulatory T cells as a distinguishing feature of colorectal tumors. J Immunol 2019; 202: 1871–84. ArticlePubMed
- 41. Di J, Liu M, Fan Y, et al. Phenotype molding of T cells in colorectal cancer by single-cell analysis. Int J Cancer 2020; 146: 2281–95. ArticlePubMed
- 42. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010; 138: 2073–87. ArticlePubMedPMC
- 43. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res 2016; 22: 813–20. ArticlePubMed
- 44. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015; 5: 43–51. ArticlePubMed
- 45. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017; 357: 409–13. ArticlePubMedPMC
- 46. Schrock AB, Ouyang C, Sandhu J, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSIhigh metastatic colorectal cancer. Ann Oncol 2019; 30: 1096–103. ArticlePubMedPDF
- 47. Klempner SJ, Fabrizio D, Bane S, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist 2020; 25: e147–59. ArticlePubMed
- 48. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017; 9: 34.ArticlePubMedPMCPDF
- 49. Merino DM, McShane LM, Fabrizio D, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer 2020; 8: e000147.ArticlePubMedPMC
- 50. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–7. ArticlePubMedPMCPDF
- 51. Parikh AR, He Y, Hong TS, et al. Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: implications for therapy. Oncologist 2019; 24: 1340–7. ArticlePubMedPMC
- 52. Lee DW, Han SW, Bae JM, et al. Tumor mutation burden and prognosis in patients with colorectal cancer treated with adjuvant fluoropyrimidine and oxaliplatin. Clin Cancer Res 2019; 25: 6141–7. ArticlePubMed
- 53. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 2018; 7: 746–56. ArticlePubMedPMC
- 54. Liu Y, Sethi NS, Hinoue T, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 2018; 33: 721–35. ArticlePubMedPMC
- 55. Hino H, Shiomi A, Kusuhara M, et al. Clinicopathological and mutational analyses of colorectal cancer with mutations in the POLE gene. Cancer Med 2019; 8: 4587–97. ArticlePubMedPMC
- 56. Domingo E, Freeman-Mills L, Rayner E, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol 2016; 1: 207–16. ArticlePubMed
- 57. Wang C, Gong J, Tu TY, Lee PP, Fakih M. Immune profiling of microsatellite instability-high and polymerase epsilon (POLE)-mutated metastatic colorectal tumors identifies predictors of response to anti-PD-1 therapy. J Gastrointest Oncol 2018; 9: 404–15. ArticlePubMedPMC
- 58. Silberman R, Steiner DF, et al. Complete and prolonged response to immune checkpoint blockade in POLE-mutated colorectal cancer. JCO Precis Oncol 2019 Jun 21 [Epub]. https://doi.org/10.1200/PO.18.00214. Article
- 59. De Sousa EM, Wang X, Jansen M, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med 2013; 19: 614–8. ArticlePubMedPDF
- 60. Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 2013; 19: 619–25. ArticlePubMedPMCPDF
- 61. Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013; 10: e1001453.ArticlePubMedPMC
- 62. Karpinski P, Rossowska J, Sasiadek MM. Immunological landscape of consensus clusters in colorectal cancer. Oncotarget 2017; 8: 105299–311. ArticlePubMedPMC
- 63. Soldevilla B, Carretero-Puche C, Gomez-Lopez G, et al. The correlation between immune subtypes and consensus molecular subtypes in colorectal cancer identifies novel tumour microenvironment profiles, with prognostic and therapeutic implications. Eur J Cancer 2019; 123: 118–29. ArticlePubMed
- 64. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity 2018; 48: 812–30. ArticlePubMedPMC
- 65. Rosenbaum MW, Bledsoe JR, Morales-Oyarvide V, Huynh TG, Mino-Kenudson M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod Pathol 2016; 29: 1104–12. ArticlePubMedPDF
- 66. Lee LH, Cavalcanti MS, Segal NH, et al. Patterns and prognostic relevance of PD-1 and PD-L1 expression in colorectal carcinoma. Mod Pathol 2016; 29: 1433–42. ArticlePubMedPMCPDF
- 67. Kim JH, Park HE, Cho NY, Lee HS, Kang GH. Characterisation of PD-L1-positive subsets of microsatellite-unstable colorectal cancers. Br J Cancer 2016; 115: 490–6. ArticlePubMedPMCPDF
- 68. Inaguma S, Lasota J, Wang Z, Felisiak-Golabek A, Ikeda H, Miettinen M. Clinicopathologic profile, immunophenotype, and genotype of CD274 (PD-L1)-positive colorectal carcinomas. Mod Pathol 2017; 30: 278–85. ArticlePubMedPDF
- 69. Eriksen AC, Sorensen FB, Lindebjerg J, et al. Programmed death ligand-1 expression in stage II colon cancer - experiences from a nationwide populationbased cohort. BMC Cancer 2019; 19: 142.ArticlePubMedPMCPDF
- 70. Li Y, He M, Zhou Y, et al. The prognostic and clinicopathological roles of PD-L1 expression in colorectal cancer: a systematic review and meta-analysis. Front Pharmacol 2019; 10: 139.ArticlePubMedPMC
- 71. Yamada R, Yamaguchi T, Iijima T, et al. Differences in histological features and PD-L1 expression between sporadic microsatellite instability and Lynch-syndrome-associated disease in Japanese patients with colorectal cancer. Int J Clin Oncol 2018; 23: 504–13. ArticlePubMedPDF
- 72. Ganesh K, Stadler ZK, Cercek A, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 2019; 16: 361–75. ArticlePubMedPMCPDF
References
Figure & Data
References
Citations
- Targeting the “tumor microenvironment”: RNA-binding proteins in the spotlight in colorectal cancer therapy
Yiwei Zhang, Yujun Zhang, Jingjing Song, Xifu Cheng, Chulin Zhou, Shuo Huang, Wentao Zhao, Zhen Zong, Lingling Yang
International Immunopharmacology.2024; 131: 111876. CrossRef - Unraveling the Role of Molecular Profiling in Predicting Treatment Response in Stage III Colorectal Cancer Patients: Insights from the IDEA International Study
Ippokratis Messaritakis, Eleni Psaroudaki, Konstantinos Vogiatzoglou, Maria Sfakianaki, Pantelis Topalis, Ioannis Iliopoulos, Dimitrios Mavroudis, John Tsiaoussis, Nikolaos Gouvas, Maria Tzardi, John Souglakos
Cancers.2023; 15(19): 4819. CrossRef - Biomarkers for Predicting Response to Personalized Immunotherapy in Gastric Cancer
Moonsik Kim, Ji Yun Jeong, An Na Seo
Diagnostics.2023; 13(17): 2782. CrossRef - Evaluation on Braking Stability of Autonomous Vehicles Running along Curved Sections Based on Asphalt Pavement Adhesion Properties
Binshuang Zheng, Xiaoming Huang, Junyao Tang, Jiaying Chen, Runmin Zhao, Zhengqiang Hong, Tao Tang, Meiling Han, Yang Yang
Journal of Advanced Transportation.2022; 2022: 1. CrossRef - Intratumoral spatial heterogeneity of tumor-infiltrating lymphocytes is a significant factor for precisely stratifying prognostic immune subgroups of microsatellite instability-high colorectal carcinomas
Minsun Jung, Ji Ae Lee, Seung-Yeon Yoo, Jeong Mo Bae, Gyeong Hoon Kang, Jung Ho Kim
Modern Pathology.2022; 35(12): 2011. CrossRef - Association of Tumor-Infiltrating Lymphocytes With Survival in Stages II and III Colorectal Cancer
Marina Vitorino, Inês Eiriz, Tiago C Tomás, Rodrigo Vicente, Ana Mendes, Ana Rita Freitas, Sofia Braga, Catarina Alves-Vale, Paula Borralho, André Ferreira, Luisa Leal da Costa
Cureus.2022;[Epub] CrossRef - Tumor Mutational Burden Predicting the Efficacy of Immune Checkpoint Inhibitors in Colorectal Cancer: A Systematic Review and Meta-Analysis
Yan Li, Yiqi Ma, Zijun Wu, Fanxin Zeng, Bin Song, Yanrong Zhang, Jinxing Li, Su Lui, Min Wu
Frontiers in Immunology.2021;[Epub] CrossRef - Genomic and transcriptomic characterization of heterogeneous immune subgroups of microsatellite instability-high colorectal cancers
Jung Ho Kim, Mi-Kyoung Seo, Ji Ae Lee, Seung-Yeon Yoo, Hyeon Jeong Oh, Hyundeok Kang, Nam-Yun Cho, Jeong Mo Bae, Gyeong Hoon Kang, Sangwoo Kim
Journal for ImmunoTherapy of Cancer.2021; 9(12): e003414. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-