E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 50(2); 2016 > Article
-
Case Study
An Adult Case of Bartter Syndrome Type III Presenting with Proteinuria - Eun Jung Cha, Won Min Hwang1, Sung-Ro Yun1, Moon Hyang Park,
-
Journal of Pathology and Translational Medicine 2016;50(2):160-164.
DOI: https://doi.org/10.4132/jptm.2015.08.31
Published online: January 11, 2016
Department of Pathology, Konyang University Hospital, Konyang University College of Medicine, Daejeon, Korea
1Division of Nephrology, Department of Internal Medicine, Konyang University Hospital, Konyang University College of Medicine, Daejeon, Korea
- Corresponding Author: Moon Hyang Park, MD Department of Pathology, Konyang University Hospital, Konyang University College of Medicine, 158 Gwanjeodong-ro, Seo-gu, Daejeon 35365, Korea Tel: +82-42-600-9280 Fax: +82-42-600-9280 E-mail: 'parkmh@hanyang.ac.kr'
• Received: June 12, 2015 • Revised: August 4, 2015 • Accepted: August 31, 2015
© 2016 The Korean Society of Pathologists/the Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Bartter syndrome (BS) I–IV is a rare autosomal recessive disorder affecting salt reabsorption in the thick ascending limb of the loop of Henle. This report highlights clinicopathological findings and genetic studies of classic BS in a 22-year-old female patient who presented with persistent mild proteinuria for 2 years. A renal biopsy demonstrated a mild to moderate increase in the mesangial cells and matrix of most glomeruli, along with marked juxtaglomerular cell hyperplasia. These findings suggested BS associated with mild IgA nephropathy. Focal tubular atrophy, interstitial fibrosis, and lymphocytic infiltration were also observed. A genetic study of the patient and her parents revealed a mutation of the CLCNKB genes. The patient was diagnosed with BS, type III. This case represents an atypical presentation of classic BS in an adult patient. Pathologic findings of renal biopsy combined with genetic analysis and clinicolaboratory findings are important in making an accurate diagnosis.
- Mild proteinuria had been incidentally detected in a 22-year-old woman during a regular health check-up 2 years prior to presentation. She visited a local clinic for a follow-up check. She took medication to treat hyperlipidemia for 1 year and did not take any other drugs including diuretics or laxatives. She was admitted to our hospital for evaluation of persistent mild proteinuria.
- The prenatal course was unremarkable. On physical examination, her height was 153 cm, her body weight was 49 kg and her blood pressure was 100/60 mm Hg. No edema was found, and normal muscle strength and reflexes were noted. The rest of the physical examination was unremarkable. The laboratory examination revealed blood urea nitrogen of 10.9 mg/dL, creatinine of 0.68 mg/dL, sodium of 137 mmol/L, potassium of 2.59 mmol/L, chloride of 94.7 mmol/L, magnesium of 2.05 mEq/L, and bicarbonate of 31.2 mEq/L. Serum levels of IgG, IgA, IgM, C3, and C4 were normal. Anti-nuclear antibody was negative. Urinalysis showed a specific gravity of 1.007, trace protein, pH 8.0, and no red blood cells. The spot urine protein/creatinine ratio was 0.95 g/g creatinine. Abdominal sonography revealed normal-sized kidneys, without nephrocalcinosis or stones. Plasma renin activity was elevated at 27.98 ng/mL/hr (normal range, 0.50 to 1.90 ng/mL/hr in a supine position), but serum aldosterone was 13.2 pg/mL within normal limits (normal range, 1 to 16 pg/mL in a supine position). The urine prostaglandin E2 level was elevated at 2,815 ng/day (normal range, 400 to 620 ng/day). These findings suggested BS or pseudo-BS/GS caused by vomiting or diuretics.
- Light microscopy of renal biopsy demonstrated 14 glomeruli, two of which were globally sclerotic. Most glomeruli appeared moderately increased in size and cellularity due to prominence of the mesangium and JGA. Five glomeruli showed marked enlargement with hyperplasia (Fig. 1A) and hypergranulosis of the JGA (Fig. 1B). The mesangium was diffusely expanded due to mild to moderate increase in cells and matrix (Fig. 1A). There was moderate tubular atrophy and interstitial fibrosis with infiltration of lymphocytes. The interlobular arteries and arterioles displayed mild to moderate intimal fibrous thickening and medial sclerosis. Immunofluorescence revealed weak positive (1+) staining for IgG, IgA, and fibrinogen and trace (+/–) of staining for C3 and lambda in the mesangium (Fig. 1C). Electron microscopy revealed mild increase in the mesangial cells and matrix with rare small mesangial electron-dense deposits and hyperplastic juxtaglomerular cells with increased electron dense renin and progranules (Fig. 1D).
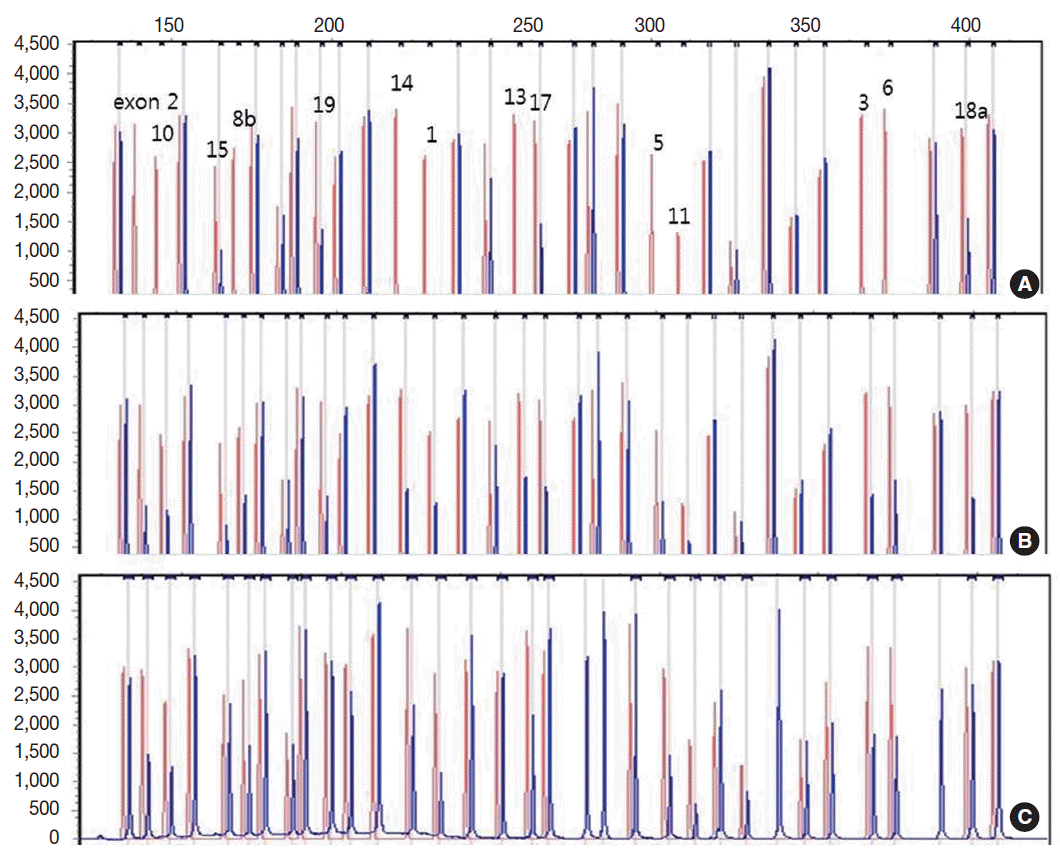
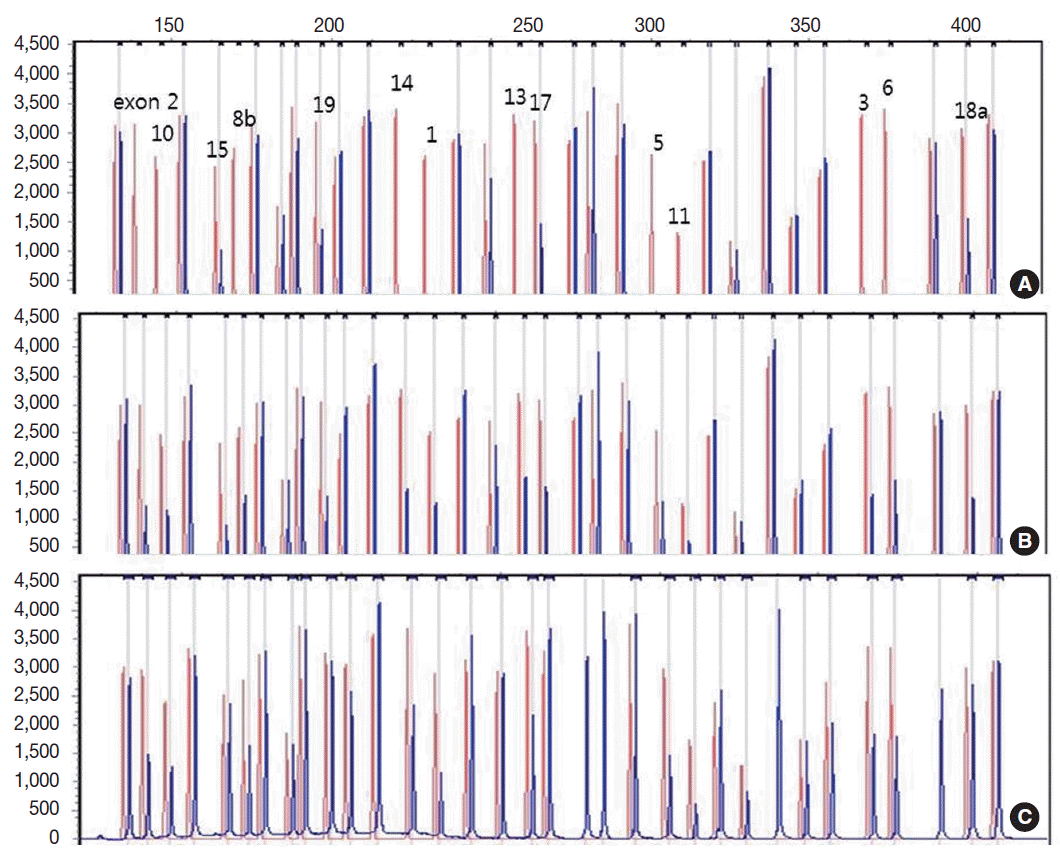
- Genetic analysis revealed a homozygous deletion of exon 1–14 and heterozygous deletion of exon 15–19 in the CLCNKB gene (Fig. 2A). Her father had heterozygous deletion of exon 1–14 (Fig. 2B), and her mother had heterozygous deletion of all examined exons of the CLCNKB gene (Fig. 2C). However, her parents displayed no renal symptoms or abnormal renal function.
CASE REPORT
- BS is an autosomal recessive genetic disorder whose primary pathogenic mechanism is defective transepithelial chloride reabsorption in the thick ascending limb of the loop of Henle. The disorder is the result of defective function of proteins responsible for transporting ions to renal cells. Five types of genetic mutations are associated with the five different forms of the disease. Type I is due to an SLC12A1 mutation encoding the bumetanide-sensitive Na+ -K+ -2Cl- cotransporter (NKCC2), and type II is the result of KCNJ1 encoding the inward rectifying K+ channel (ROMK). Type III or classic BS results from a mutation of CLCNKB, encodes for the kidney-specific basolateral chloride channel (CIC-Kb). The CIC-Kb protein is required to ensure the exit of chloride (Cl-) on the basolateral side. Impaired CICKb function reduces Cl- efflux and decreases Na-K-Cl reabsorption through NKCC2 by modifying the transepithelial voltage gradient and causing salt loss in urine [4]. Type IV is due to mutations of BSND encoding barttin (the β-subunit of the basolateral chloride channel). Finally, type V, with the features of autosomal dominant hypocalcemia, is caused by gain-of-function mutations of the calcium-sensing receptor [3,5,8,9]. Types I, II, and IV are classified as antenatal BS.
- Renal calcium handling can be used to differentiate between classic BS and GS. Hypomagnesemia is considered a well-defined characteristic of GS but it also affects a considerable number of patients with BS. The most definitive diagnostic method of BS is molecular analysis [10,11]. The patient in the current case had no symptoms in the neonatal period or in childhood. This patient had no symptoms of polyuria, dehydration, or polydipsia except for nocturia and presented with mild proteinuria and mild hypokalemic metabolic alkalosis at 22 years of age. Clinically, this patient’s diagnosis raised a suspicion of GS rather than BS, but deletion of the CLCNKB gene in the genetic analysis confirmed type III BS.
- Although proteinuria is not a classic symptom in BS, it has been a presenting symptom in cases associated with other glomerular diseases, such as focal segmental glomerulosclerosis, C1q nephropathy or immune complex glomerulopathy [12-15]. Our patient had mild proteinuria for 2 years. The presence of mild to moderate mesangial proliferation in the renal biopsy, in addition to the weak positive immunofluorescence staining for IgA in the mesangium pointed to the possibility of mild IgA nephropathy coexistent with BS. Electron microscopy revealed rare small paramesangial electron dense deposits.
- Renal changes in BS are probably caused by stimulation of the renin-angiotensin axis and activation of transforming growth factor β (TGF-β) [16]. Laboratory studies have suggested that exposure of mesangial cells to angiotensin II results in proliferation, hypertrophy, and TGF-β production [13]. Although patients with BS have high angiotensin II level and activation of the renin-angiotensin axis, they also demonstrate normo/hypotension, reduced peripheral resistance, and hyporesponsiveness to vasopressor agents. Patients with BS and GS, as well as heterozygous carriers of both disorders, have lower blood pressure than the general population [2]. In addition to volume depletion, another possible contributor to the lower blood pressure in BS is increased renal release of vasodilator prostaglandins E2 (PGE2). The increased renal production of PGE2 results from impaired entry of sodium chloride into the macula densa cells at the end of the thick ascending limb of the loop of Henle, which increases the expression of cyclooxygenase 2 [17]. Therefore, these patients do not develop hypertension or associated complications such as cardiovascular remodeling and atherogenesis [18]. The cause of renal dysfunction in patients with BS is unclear.
- The histologic and molecular features of BS have been well described in the literature [3,5,6], and histologic findings of BS consistently show juxtaglomerular hyperplasia, interstitial fibrosis and nephrocalcinosis. Juxtaglomerular hyperplasia is a characteristic finding but is not a necessary finding for diagnosis. Juxtaglomerular hyperplasia can be associated with a variety of causes, such as chronic hypovolemic states with hypokalemia due to chronic vomiting or laxative abuse, familial chloride diarrhea, or cystinosis [4]. The ingestion of diuretics is perhaps the most common cause of chronic hypokalemic alkalosis. For an accurate diagnosis, other causes must be rule out through careful history taking, plasma and urine electrolyte measurement, or various diuretic screening methods [19].
- Accounting for the patient’s clinical, laboratory, and pathological findings of BS, in conjunction with the results of genetic analysis of both the patient and her parents, the patient was diagnosed with type III BS. This case shows the importance of renal biopsy and molecular analysis in delineating the cause of an atypical presentation of classic BS in an adult patient.
DISCUSSION
-
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Notes
Acknowledgments
Fig. 1.Renal biopsy in a patient with Bartter syndrome with IgA nephropathy. (A) Glomerulus shows hyperplasia of juxtaglomerular cells with increased mesangial cells and matrix (periodic acid-Schiff). (B) Glomerulus shows hyperplasia and hypergranulosis of juxtaglomerular cells (Jones’ methenamine silver, × 1,000). (C) Immunofluorescence reveals weak (1+) staining for IgA in the mesangium. (D) Electron micrograph of the juxtraglomerular apparatus shows abundant progranules and mature renin granules (Hitach HT7700 EM, × 6,000).
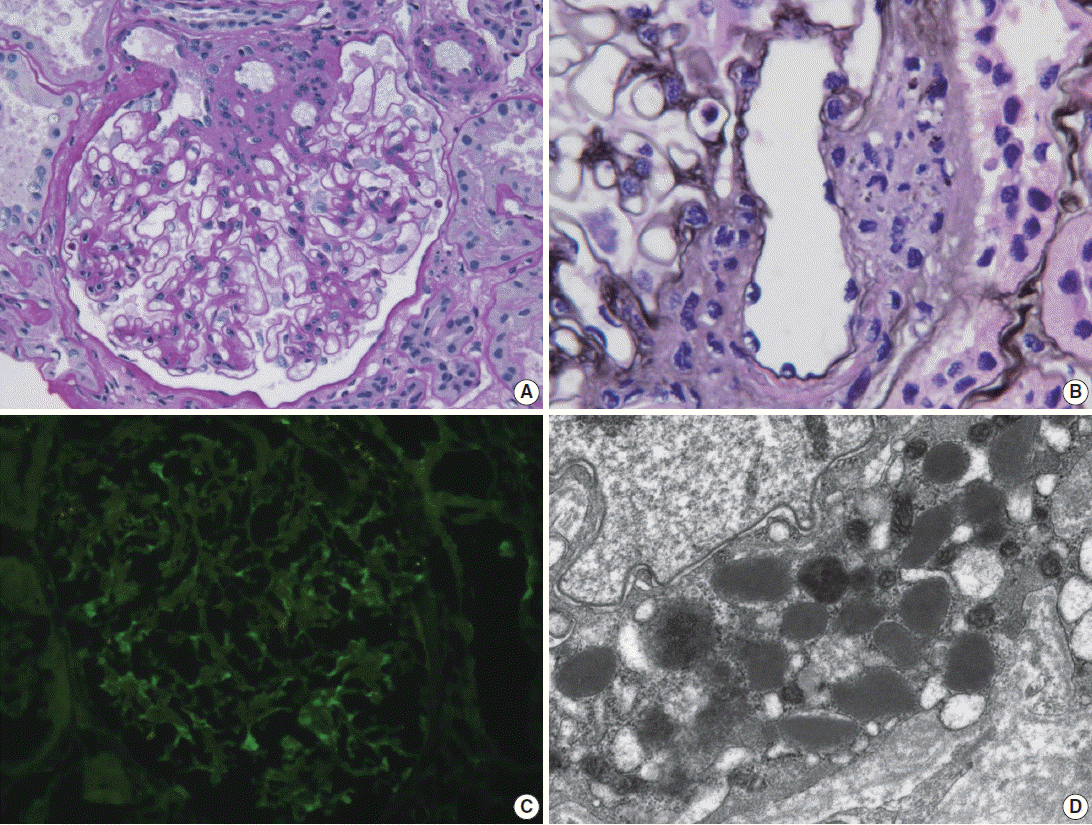
Fig. 2.Capillary electrophoretic pattern of the multiplex ligation-dependent probe amplification products of the patient’s family. (A) The patient has a large homozygous deletion and a large heterozygous deletion. (B) Her father has a heterozygous deletion of exon 1–14. (C) Her mother has a heterozygous deletion of all examined exons of the CLCNKB gene.
- 1. Bartter FC, Pronove P, Gill JR Jr, Maccardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. Am J Med 1962; 33: 811–28. ArticlePubMed
- 2. Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 2008; 40: 592–9. ArticlePubMedPMC
- 3. Lee BH, Cho HY, Lee H, et al. Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant 2012; 27: 1516–21. ArticlePubMed
- 4. Naesens M, Steels P, Verberckmoes R, Vanrenterghem Y, Kuypers D. Bartter’s and Gitelman’s syndromes: from gene to clinic. Nephron Physiol 2004; 96: p65–78. ArticlePubMedPDF
- 5. Xiumin W, Zheng S, Meichun X, Junfen F, Li L. A Chinese girl with Bartter syndrome type III due to a novel mutation and/or single nucleotide polymorphisms (SNPs) in CLCNKB gene. Iran J Pediatr 2013; 23: 89–94. PubMedPMC
- 6. García Castaño A, Pérez de Nanclares G, Madariaga L, et al. Genetics of type III Bartter syndrome in Spain, proposed diagnostic algorithm. PLoS One 2013; 8: e74673. Article
- 7. Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 1997; 17: 171–8. ArticlePubMed
- 8. Bhat YR, Vinayaka G, Sreelakshmi K. Antenatal Bartter syndrome: a review. Int J Pediatr 2012; 2012: 857136.ArticlePubMedPMCPDF
- 9. Krämer BK, Bergler T, Stoelcker B, Waldegger S. Mechanisms of disease: the kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat Clin Pract Nephrol 2008; 4: 38–46. ArticlePubMed
- 10. Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis 2008; 3: 22.ArticlePubMedPMC
- 11. Nakhoul F, Nakhoul N, Dorman E, Berger L, Skorecki K, Magen D. Gitelman’s syndrome: a pathophysiological and clinical update. Endocrine 2012; 41: 53–7. ArticlePubMed
- 12. Su IH, Frank R, Gauthier BG, et al. Bartter syndrome and focal segmental glomerulosclerosis: a possible link between two diseases. Pediatr Nephrol 2000; 14: 970–2. ArticlePubMed
- 13. Sardani Y, Qin K, Haas M, Aronson AJ, Rosenfield RL. Bartter syndrome complicated by immune complex nephropathy: case report and literature review. Pediatr Nephrol 2003; 18: 913–8. ArticlePubMed
- 14. Hanevold C, Mian A, Dalton R. C1q nephropathy in association with Gitelman syndrome: a case report. Pediatr Nephrol 2006; 21: 1904–8. ArticlePubMed
- 15. Lee SE, Han KH, Jung YH, et al. Renal transplantation in a patient with Bartter syndrome and glomerulosclerosis. Korean J Pediatr 2011; 54: 36–9. ArticlePubMedPMC
- 16. Yamamoto T, Noble NA, Cohen AH, et al. Expression of transforming growth factor-beta isoforms in human glomerular diseases. Kidney Int 1996; 49: 461–9. ArticlePubMed
- 17. Kömhoff M, Reinalter SC, Gröne HJ, Seyberth HW. Induction of microsomal prostaglandin E2 synthase in the macula densa in children with hypokalemic salt-losing tubulopathies. Pediatr Res 2004; 55: 261–6. ArticlePubMed
- 18. Pagnin E, Davis PA, Semplicini A, Calò LA. The search for a link between inflammation and hypertension: contribution from Bartter’s/Gitelman’s syndromes. Nephrol Dial Transplant 2006; 21: 2340–2. Article
- 19. Colussi G, Rombolà G, Airaghi C, De Ferrari ME, Minetti L. Pseudo-Bartter’s syndrome from surreptitious diuretic intake: differential diagnosis with true Bartter’s syndrome. Nephrol Dial Transplant 1992; 7: 896–901. ArticlePubMed
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Bartter syndrome with multiple renal and liver cysts: a case report
Yemei He, Yue Zhou, Weihua Wu, Yue Chen, Santao Ou
International Urology and Nephrology.2022; 55(1): 225. CrossRef - Bartter’s syndrome: clinical findings, genetic causes and therapeutic approach
Flavia Cristina Carvalho Mrad, Sílvia Bouissou Morais Soares, Luiz Alberto Wanderley de Menezes Silva, Pedro Versiani dos Anjos Menezes, Ana Cristina Simões-e-Silva
World Journal of Pediatrics.2021; 17(1): 31. CrossRef - Association of Adult-Onset Bartter Syndrome With Undifferentiated Connective Tissue Disorder
Nida Saleem, Humaira Nasir, Danyal Hassan, Momena Manzoor
Cureus.2021;[Epub] CrossRef - Acquired autoimmune Bartter syndrome in a patient with primary hypothyroidism
Noreen Nasir, Deepali Mohanty, Arun Kumar Pande, Dhanita Khanna, Kavita Vishvakarma, Latika Gupta
Rheumatology International.2021; 43(3): 567. CrossRef - A novel mutation associated with Type�III Bartter syndrome: A report of five cases
Yanhan Li, Chengcheng Wu, Jie Gu, Dong Li, Yanling Yang
Molecular Medicine Reports.2019;[Epub] CrossRef - Pathophysiology of antenatal Bartterʼs syndrome
Martin Kömhoff, Kamel Laghmani
Current Opinion in Nephrology and Hypertension.2017; 26(5): 419. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-