E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 47(1); 2013 > Article
-
Case Study
Peripheral Primitive Neuroectodermal Tumor with Osseous Component of the Small Bowel Mesentery: A Case Study - Joon Mee Kim, Young Chae Chu, Chang Hwan Choi, Lucia Kim, Suk Jin Choi, In Suh Park, Jee Young Han, Kyung Rae Kim1, Yoon-La Choi2, Taeeun Kim3
-
Korean Journal of Pathology 2013;47(1):77-81.
DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.1.77
Published online: February 25, 2013
Department of Pathology, Inha University Hospital, Inha University School of Medicine, Incheon, Korea.
1Department of Surgery, Inha University Hospital, Inha University School of Medicine, Incheon, Korea.
2Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
3Department of Pathology, Gachon University of Medicine and Science, Incheon, Korea.
- Corresponding Author: Young Chae Chu, M.D. Department of Pathology, Inha University Hospital, Inha University School of Medicine, 27 Inhang-ro, Jung-gu, Incheon 400-711, Korea. Tel: +82-32-890-3984, Fax: +82-32-890-3464, 'ycchu@inha.ac.kr'
© 2013 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- A case of peripheral primitive neuroectodermal tumor of the small bowel mesentery with osseous component is reported. A 23-year-old man was admitted to our hospital because of acute severe abdominal pain. Abdominal computed tomography revealed a large solid and cystic, oval shaped mass, measuring 11.0×6.0 cm in the pelvic cavity. Histologically the resected lesion consisted of sheets of undifferentiated small round cells forming Homer-Wright rosettes and perivascular pseudorosettes, and showed areas of osteoid and bone formation. Immunohistochemical studies revealed that tumor cells expressed positivity against CD99 (MIC2), CD57, neuron-specific enolase, and vimentin. Fluorescence in situ hybridization study revealed Ewing sarcoma breakpoint region 1 (EWSR1) gene rearrangement on chromosome 22q12. To the authors' knowledge this is the first documentation of a peripheral neuroectodermal tumor with osteoid and bone formation of the small bowel mesentery.
- A 23-year-old man was referred to Inha University Hospital due to severe abdominal pain. Computed tomography revealed an ovoid solid and cystic tumor in the pelvic cavity, measuring 11.0×6.0 cm. Emergency surgery was performed, and a large ruptured mass was found in the jejunal mesentery, 1 cm from the ligament of Treitz. The tumor involved the jejunal wall, and another mass the size of a thumb was detected within the porta hepatis. Disseminated miliary nodules were present in the greater omentum, the right colonic gutter, and the pelvic peritoneum. Segmental resection of the small intestine and omentectomy were performed. Postoperative laboratory examination revealed normal levels of serum lactate dehydrogenase, cancer antigen 125, and carcinoembryonic antigen, and postoperative positron emission tomography computed tomography was normal. After operation, chemotherapy (vincristine, ifosfamide, etoposide, and doxorubicin) was performed. One year after the operation, a 10 cm, locally recurrent mesenteric mass was resected.
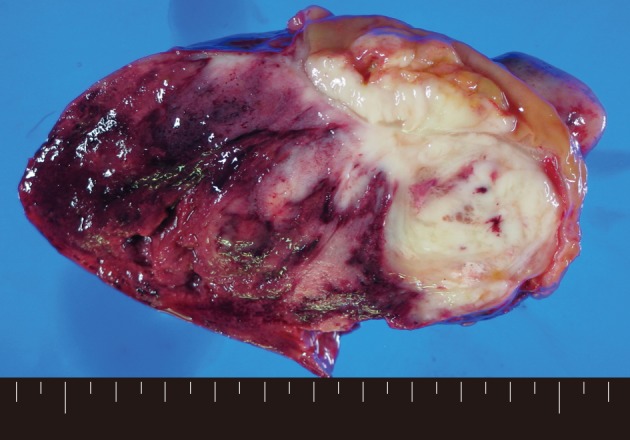
- Macroscopically, a tumor measuring 12.0×8.0×7.5 cm was located in the mesentery of jejunum. Rupture was seen in the surface of tumor. The tumor showed a whitish-pink solid cut surface with foci of hemorrhage, extensive necrosis, and myxoid change. The jejunal wall was involved directly by the tumor and contained focal mucosal ulceration. The cut surface was white-gray and dense (Fig. 1).
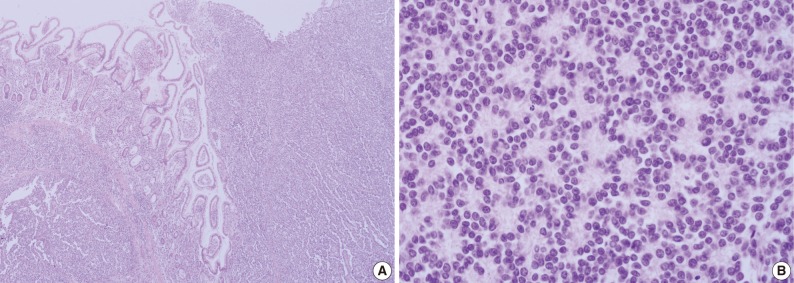
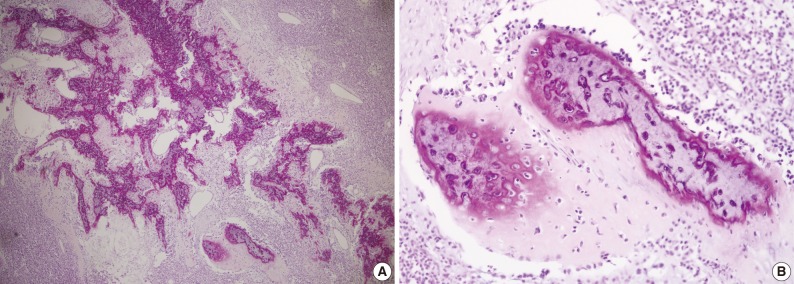
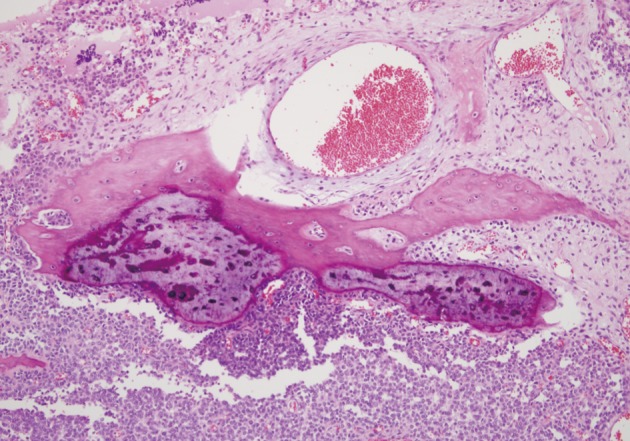
- Microscopically, the entire wall of the jejunum contained tumor cells which also proliferated within the mesentery. The mucosa of the jejunum was involved by the tumor with surface erosion (Fig. 2A). Small round cells containing uniform vesicular, round or oval nuclei, scanty clear, or eosinophilic cytoplasms and indistinct cytoplasmic borders comprised the tumor in sheet or lobule formation (Fig. 2B). Often, intermingled spindle-shaped cells were found. Though some cells had a single prominent nucleolus, most cells had indistinct nucleoli. Well-formed Homer-Wright rosettes were frequently observed (Fig. 2B). Flexner-Wintersteiner rosettes did not exist, but perivascular pseudorosettes were occasionally seen. The tumor contained concentrated mild desmoplastic stromal changes. Mitotic figures were present in approximately 35 per 10 high-powered fields. Areas of production of osseous matrix with calcification and partly well-formed bony trabeculae were observed (Fig. 3A). The osteogenic area was not more than 1% of the tumor. Although cytologically malignant cells were admixed with this osteogenic area, benign metaplastic bone formation was suspected, because the atypical cells were present only in the peripheral portion of the osteoid (Fig. 3B) and positive for CD99. The osteogenic area, in contrast, was CD99 negative. The pathologic findings of the metastatic lesions of the omental mass were identical to those of the primary mesenteric mass. Cytoplasmic glycogen was undetected in the tumor cells when pretreated with and without diastase in periodic acid-Schiff reactions. No cytoplasmic reactivity was found with Grimelius and Masson-Fontana staining.
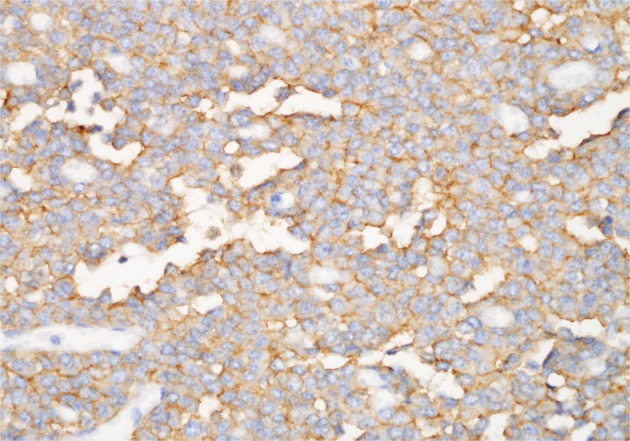
- Positive CD99 (MIC2), CD57, and neuron-specific enolase identified many tumor cells immunohistochemically (Fig. 4), whereas positive vimentin and neurofilament results were also present in some cells. Synaptophysin, chromogranin, desmin, cytokeratin, S-100, CD117, and leukocyte common antigen were all absent in the tumor cells.
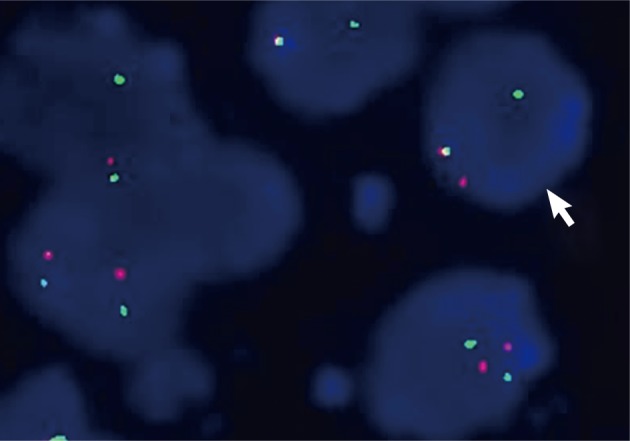
- Using dual color break-apart Ewing's sarcoma probing, fluorescence in situ hybridization (FISH) was performed on sections of the tissues that were formalin-fixed and paraffin embedded (Vysis, Downers Grove, IL, USA) with a mixture of 2 FISH DNA probes. The first was a 500-kb probe, labeled in the spectrum orange, and flanking the 5' side of the Ewing sarcoma breakpoint region 1 (EWSR1) gene. The second probe, which flanked the 3' end of the EWSR1 gene, was a 1,100-kb probe, utilizing a spectrum green label. Introns 7 through 10, used as restrictions within the EWSR1 gene, were the known break points. FISH showed a split signal pattern (one green and one orange) in interphase nuclei which was indicative of a EWSR1 gene rearrangement (Fig. 5). A pPNET of the small bowel mesentery diagnosis was ascribed to the lesion, given these results.
- The recurrent tumor resected one year after surgery, revealed similar histologic features: a typical small round cell tumor with rosette formation and metaplastic bone formation (Fig. 6). The bony islands were more mature than the primary tumor.
CASE REPORT
- The entire body is vulnerable to peripheral primitive neuroectodermal tumor invasion. The primary sites of pPNET are, indescending frequency, the chest wall, pelvis, retroperitoneum, abdomen, limb, and neck.10 In viscera, distinct cases of pPNET have been studied.3-6 However, in the English literature, only one case of pPNET of the mesentery was reported with perforation at presentation as was presented in our case study.4 pPNET prognosis is poor despite combined surgical, chemotherapeutic, and irradiation therapies. Only 25% of patients with tumors greater than 5 cm survive to 24 months according to Kushner et al.10 Histologically, Homer-Wright or Flexner-Wintersteiner rosettes and perivascular pseudorosettes may form from undifferentiated small round cells which constitute pPNET. Fibrosarcoma or malignant peripheral nerve sheath tumors, small cell undifferentiated carcinomas, and carcinoid tumors may resemble some areas within the lesions. It is known that tumors of neural crest origin can show bidirectional or multidirectional differentiation.7-9
- Additionally, glial, ependymal, cartilaginous, and epithelial elements, though rare, have been found associated within pPNET.7-9 Hachitanda et al.11 reported a case of pPNET with epithelial and glial differentiation, and they suggested that the neoplastic neuroectodermal tissue can display a spectrum of differentiation. Although there has been no report of pPNET showing osteoid and bone production, it is thought that osteogenesis is a kind of differentiation of the tumor. Its prognostic implication is uncertain. Although several cases of bone and/or cartilage forming sarcomas have been reported in the literature,12-15 bone-forming pPNET has not.
- Most authors agree that a useful tool in diagnosing pPNET immunohistochemically is CD99 (MIC2), which recognizes a 30/32 kDa surface glycoprotein.16 This marker is found in more than 90% of pPNET cases. Yet, many tumors, such as malignant lymphoma, leukemia, gastrointestinal stromal tumor, and small cell carcinoma, may demonstrate CD99 expression.17-20 Regarding pediatric malignant lymphoma and leukemia of T-cell lineage, Riopel et al.17 reported that CD99 expression was not uncommon.
- The most objective diagnostic tool for pPNET is now considered to be karyotypic analysis for t(11;22)(q24;q12) translocation.2,16 This translocation occurs in more than 87% of the pPNET-Ewing's sarcoma cases. The detection of EWS/FLI-1 chimeric mRNA originating from the t(11;22)(q24;q12) translocation of the pPNET-Ewing's sarcoma family, facilitated by reverse transcription-polymerase chain reactions, have been reported in recent studies.2
- Other small round cell tumors, including malignant lymphoma, leukemia (granulocytic sarcoma), rhabdomyosarcoma, leiomyosarcoma, gastrointestinal stromal tumor, desmoplastic small round cell tumor, malignant mesothelioma, undifferentiated carcinoma, small cell carcinoma, and conventional neuroblastoma offer a differential diagnosis of the current lesion being discussed. Through histological, histochemical, immunohistochemical and molecular methods, the lesion was meticulously examined to maintain distinction. Immunohistochemical staining with desmin, smooth muscle actin, CD34, cytokeratin, leukocyte common antigen, CD117, and CD99 were used to exclude the diagnosis of other small round cell tumors and gastrointestinal stromal tumors. In addition, chromosomal rearrangements involving the EWSR1 gene on chromosome 22q12 was detected by FISH, which was a strong supportive finding for pPNET. Most of the mass at the primary site was found in the mesentery of the jejunum. Direct invasion of the jejunal wall was also present, yet despite the large size of the tumor (12.0×8.0 cm), the degree of involvement of the mucosa of the jejunum was relatively limited. A final diagnosis of pPNET of the jejunal mesentery was applied to this tumor.
- In conclusion, we have reported the first case of pPNET with osteoid and bone formation, arising from the mesentery of the small bowel with rupture at onset. The rupture was considered to be caused by local ischemic change, necrosis, and massive hemorrhage. Intraabdominal pPNET may present with acute abdomen and may reveal osteoid and bone formation, which leads to a high degree of importance for both surgeons and pathologists to consider.
DISCUSSION
Acknowledgments
Acknowledgments
- 1. Dehner LP. Primitive neuroectodermal tumor and Ewing's sarcoma. Am J Surg Pathol 1993; 17: 1–13. PMID: 8383465. ArticlePubMed
- 2. Kawauchi S, Fukuda T, Miyamoto S, et al. Peripheral primitive neuroectodermal tumor of the ovary confirmed by CD99 immunostaining, karyotypic analysis, and RT-PCR for EWS/FLI-1 chimeric mRNA. Am J Surg Pathol 1998; 22: 1417–1422. PMID: 9808135. ArticlePubMed
- 3. Bala M, Maly A, Remo N, Gimmon Z, Almogy G. Peripheral primitive neuroectodermal tumor of bowel mesentery in adults. Isr Med Assoc J 2006; 8: 515–516. PMID: 16889176. PubMed
- 4. Horie Y, Kato M. Peripheral primitive neuroectodermal tumor of the small bowel mesentery: a case showing perforation at onset. Pathol Int 2000; 50: 398–403. PMID: 10849329. ArticlePubMed
- 5. Tokudome N, Tanaka K, Kai MH, Sueyoshi K, Matsukita S, Setoguchi T. Primitive neuroectodermal tumor of the transverse colonic mesentery defined by the presence of EWS-FLI1 chimeric mRNA in a Japanese woman. J Gastroenterol 2002; 37: 543–549. PMID: 12162413. ArticlePubMed
- 6. Balasubramanian B, Dinakarababu E, Molyneux AJ. Primary primitive neuroectodermal tumour of the small bowel mesentery: case report. Eur J Surg Oncol 2002; 28: 197–198. PMID: 11884059. ArticlePubMed
- 7. Shuangshoti S. Primitive neuroectodermal (neuroepithelial) tumour of soft tissue of the neck in a child: demonstration of neuronal and neuroglial differentiation. Histopathology 1986; 10: 651–658. PMID: 2426177. ArticlePubMed
- 8. Shinoda M, Tsutsumi Y, Hata J, Yokoyama S. Peripheral neuroepithelioma in childhood: immunohistochemical demonstration of epithelial differentiation. Arch Pathol Lab Med 1988; 112: 1155–1158. PMID: 3178431. PubMed
- 9. Karcioglu Z, Someren A, Mathes SJ. Ectomesenchymoma: a malignant tumor of migratory neural crest (ectomesenchyme) remnants showing ganglionic, schwannian, melanocytic and rhabdomyoblastic differentiation. Cancer 1977; 39: 2486–2496. PMID: 872048. ArticlePubMed
- 10. Kushner BH, Hajdu SI, Gulati SC, Erlandson RA, Exelby PR, Lieberman PH. Extracranial primitive neuroectodermal tumors: the Memorial Sloan-Kettering Cancer Center experience. Cancer 1991; 67: 1825–1829. PMID: 1848468. ArticlePubMed
- 11. Hachitanda Y, Tsuneyoshi M, Enjoji M, Nakagawara A, Ikeda K. Congenital primitive neuroectodermal tumor with epithelial and glial differentiation: an ultrastructural and immunohistochemical study. Arch Pathol Lab Med 1990; 114: 101–105. PMID: 2294862. PubMed
- 12. Song JS, Gardner JM, Tarrant WP, et al. Dedifferentiated liposarcoma with peculiar meningothelial-like whorling and metaplastic bone formation. Ann Diagn Pathol 2009; 13: 278–284. PMID: 19608088. ArticlePubMed
- 13. Macarenco RS, Erickson-Johnson M, Wang X, Jenkins RB, Nascimento AG, Oliveira AM. Cytogenetic and molecular cytogenetic findings in dedifferentiated liposarcoma with neural-like whorling pattern and metaplastic bone formation. Cancer Genet Cytogenet 2007; 172: 147–150. PMID: 17213023. ArticlePubMed
- 14. Weidner N. Atypical tumor of the mediastinum: epithelioid hemangioendothelioma containing metaplastic bone and osteoclastlike giant cells. Ultrastruct Pathol 1991; 15: 481–488. PMID: 1755105. ArticlePubMed
- 15. Bhagavan BS, Dorfman HD. The significance of bone and cartilage formation in malignant fibrous histiocytoma of soft tissue. Cancer 1982; 49: 480–488. PMID: 6277450. ArticlePubMed
- 16. Scotlandi K, Serra M, Manara MC, et al. Immunostaining of the p30/32MIC2 antigen and molecular detection of EWS rearrangements for the diagnosis of Ewing's sarcoma and peripheral neuroectodermal tumor. Hum Pathol 1996; 27: 408–416. PMID: 8617485. ArticlePubMed
- 17. Riopel M, Dickman PS, Link MP, Perlman EJ. MIC2 analysis in pediatric lymphomas and leukemias. Hum Pathol 1994; 25: 396–399. PMID: 8163272. ArticlePubMed
- 18. Menasce LP, Banerjee SS, Beckett E, Harris M. Extra-medullary myeloid tumour (granulocytic sarcoma) is often misdiagnosed: a study of 26 cases. Histopathology 1999; 34: 391–398. PMID: 10231412. ArticlePubMed
- 19. Lumadue JA, Askin FB, Perlman EJ. MIC2 analysis of small cell carcinoma. Am J Clin Pathol 1994; 102: 692–694. PMID: 7524308. ArticlePubMed
- 20. Shidham VB, Chivukula M, Gupta D, Rao RN, Komorowski R. Immunohistochemical comparison of gastrointestinal stromal tumor and solitary fibrous tumor. Arch Pathol Lab Med 2002; 126: 1189–1192. PMID: 12296756. ArticlePubMedPDF
References
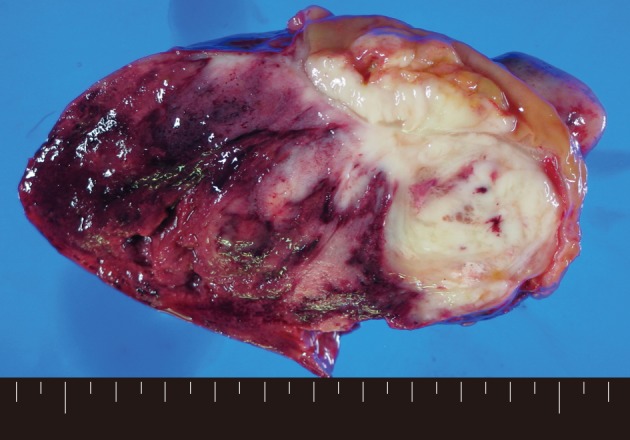
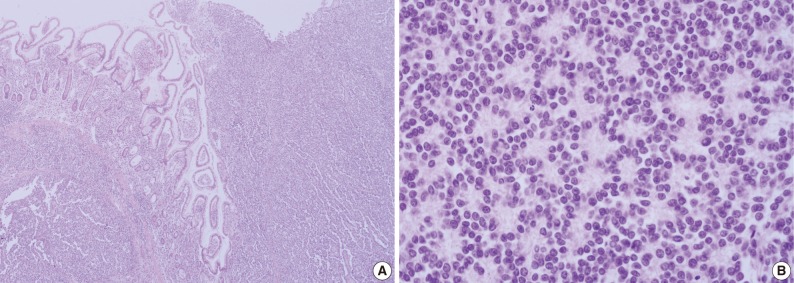
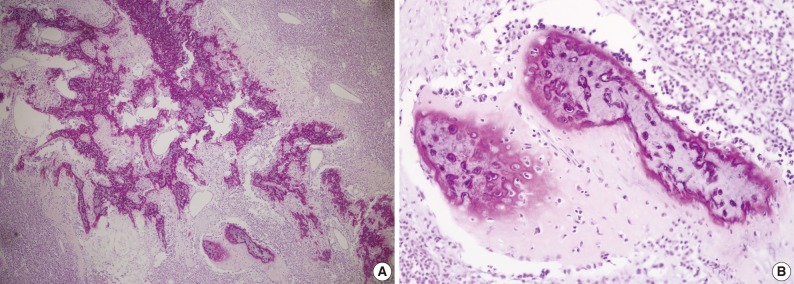
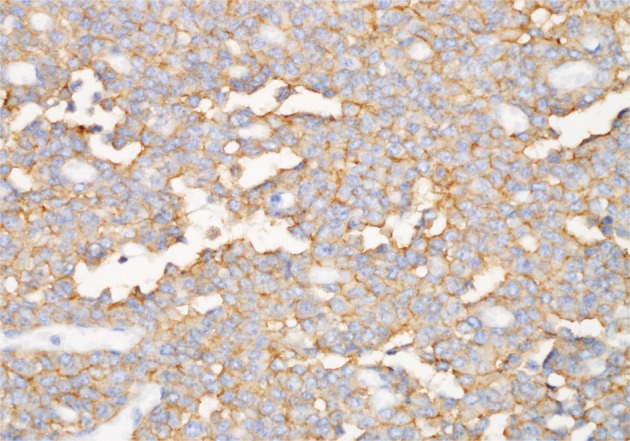
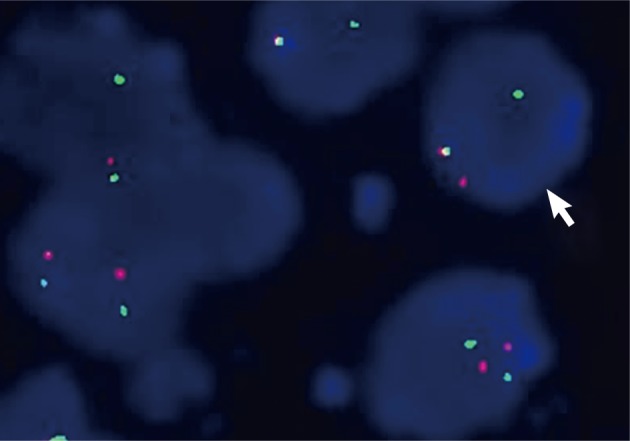
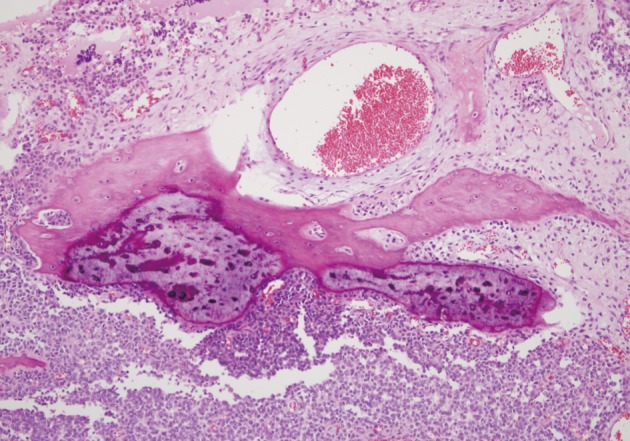
Figure & Data
References
Citations
- Primary Ewing’s sarcoma in a small intestine – a case report and review of the literature
Andrej Kolosov, Audrius Dulskas, Kastytis Pauza, Veslava Selichova, Dmitrij Seinin, Eugenijus Stratilatovas
BMC Surgery.2020;[Epub] CrossRef - Case report and literature review of Ewing's sarcoma in the gastrointestinal tract
Christopher Bong, Iain Thomson, Guy Lampe
Surgical Practice.2018; 22(2): 84. CrossRef - Pediatric Ewing’s Sarcoma/Primitive Neuroectodermal Tumor (ES/PNET) Developed in the Small Intestine: A Case Report
You Sun Kim, Hye Min Moon, Kyu Sang Lee, Young Suk Park, Hyun-Young Kim, Ji Young Kim, Jin Min Cho, Hyoung Soo Choi
Clinical Pediatric Hematology-Oncology.2017; 24(2): 162. CrossRef - Huge peripheral primitive neuroectodermal tumor of the small bowel mesentery at nonage: A case report and review of the literature
Zhe Liu, Yuan-Hong Xu, Chun-Lin Ge, Jin Long, Rui-Xia Du, Ke-Jian Guo
World Journal of Clinical Cases.2016; 4(9): 306. CrossRef - Primary primitive neuroectodermal tumor arising in the mesentery and ileocecum: A report of three cases and review of the literature
LIBO PENG, LIMIN YANG, NAN WU, BO WU
Experimental and Therapeutic Medicine.2015; 9(4): 1299. CrossRef - Une curieuse tumeur digestive à cellules rondes
Alia Zehani, Ines Chelly, Beya Chelly, Jean-Michel Coindre, Slim Haouet, Nidhameddine Kchir
Annales de Pathologie.2014; 34(2): 104. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-